Immuunijärjestelmä: luonnolliset tappajasolut
Luonnolliset tappajasolut voivat tuhota SARS-CoV-2-viruksen infektoimia soluja, mutta vakavaa COVID-19-infektiota sairastavilla NK-solujen antama immuunivaste toimii puutteellisesti.
Writkowski et.al havaitsi, että vaikeaa COVID-19-tautia sairastavien verenkierrossa on normaalia korkeampi pitoisuus anti-inflammatorisia molekyylejä, jotka vaikuttavat kasvutekijä-β1:een (TGF-β1). Tämä vaikutus liittyy NK-solujen heikentyneeseen antiviraaliseen immuunivasteeseen, mikä heikentää immuunijärjestelmän puolustusta SARS-CoV-2-virusta vastaan.
RNA-sekvensointianalyysissä NK-solut osoittivat erityispiirteitä COVID-19-potilailla terveisiin verrattuna. Proinflammatoriset sytokiinivälitteiset signalointireitit vahvistuivat merkittävästi. Epätavallinen CD56dimCD16neg NK-solupopulaatio laajeni COVID-19-potilaiden PBMC-soluissa taudin vakavuudesta riippumatta, ja siihen liittyi NK-solujen sytotoksisuuden heikkeneminen. NK-solupopulaatio kuitenkin normalisoitui nopeasti samalla kun epätavanomaiset CD56dimCD16neg NK-solut katosivat ja NK-solujen sytotoksisuus palautui potilailla, joilla oli lievä COVID-19, mutta NK-solujen sytotoksisuus palautui hitaasti potilailla, joilla oli vakava COVID-19.
Luonnollisten tappajasolujen sytotoksisuuden heikkeneminen altistaa vakavammalle COVID-19-infektiolle, mutta mitä nämä luonnolliset tappajasolut, eli NK-solut ovat?
Johdanto
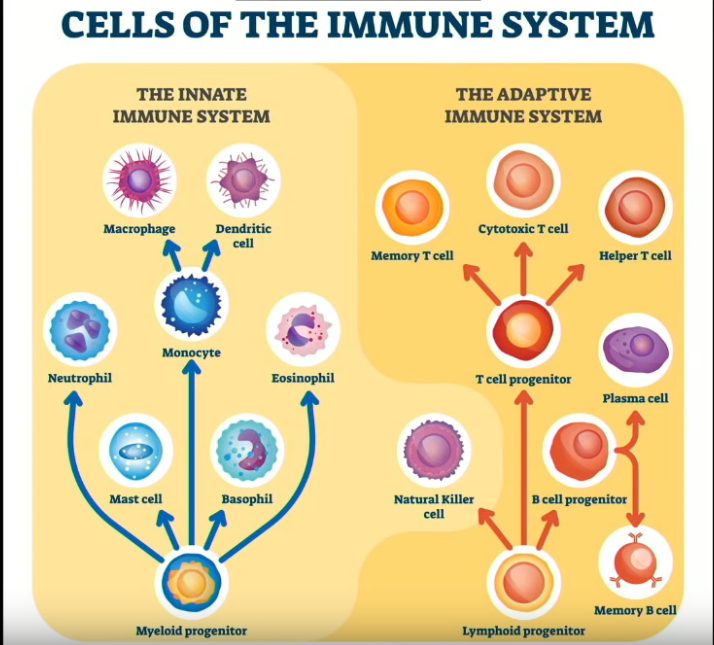
Luonnolliset tappajasolut (Natural Killer Cells) ovat T -ja B -solujen perheeseen kuuluvia yhteisestä kantasolusta kehittyviä lymfosyyttejä. Luontaisen immuunijärjestelmän soluina NK-solut luokitellaan ryhmän I synnynnäisiksi lymfosyyteiksi (ILC).
Luonnolliset tappajasolut reagoivat nopeasti patologisiin uhkiin. NK-solut tunnetaan erityisesti virustartunnan saaneiden solujen tuhoamisesta sekä syövän varhaisten merkkien havaitsemisesta ja kontrolloimisesta.
Erikoistuneita NK-soluja on myös istukassa ja niillä on tärkeä rooli hedelmöittymisessä ja raskauden aikana. NK-solut havaittiin niiden kyvystä tappaa pahanlaatuisia soluja ilman erillistä aktivointia. Sytotoksisista T-soluista poiketen NK-solut eivät tarvitse aktivointia antigeeniä esittäviltä soluilta.
NK-solut erittävät sytokiinejä: interferoni-gamma[IFNy] ja tuumorinekroositekijä-alfa[TNFa], jotka vaikuttavat immuunivastetta tehostaviin immuunisoluihin, kuten makrofageihin ja dendriittisoluihin.
NK-solut ovat jatkuvasti yhteydessä muihin soluihin. Luonnollisen tappajasolun immuunivasteen käynnistymiseen vaikuttaa aktivoivien ja estävien reseptorien signaalien tasapaino NK-solun pinnalla.
Aktivoivat reseptorit tunnistavat molekyylejä, jotka ilmentyvät pahanlaatuisten syöpäsolujen ja sairastuneiden solujen pinnalla käynnistäen NK -solun sytotoksisen immuunivasteen. Estävät (inhibitoivat) reseptorit toimivat NK -solujen varmistusmekanismina, joka estää NK-soluja tuhoamasta terveitä soluja.
Useimmat terveet solut ilmentävät pinnallaan MHC I-reseptoreita. Nämä toimivat tunnisteina, joiden avulla immuunijärjestelmä erottaa omat solut vieraista taudinaiheuttajista. NK -solun pinnalla olevat estävät reseptorit tunnistavat MHC I:n. Tämä ”sammuttaa” NK -solun immuunivasteen estäen terveen solun tappamisen.
Syöpäsolut ja viruksen infektoimat solut menettävät usein MHC I-reseptorit, jolloin ne altistuvat NK-solujen sytotoksiselle immuunivasteelle. Havaitessaan infektoituneen solun NK-solu vapauttaa perforiinia ja grantsyymejä sisältäviä sytotoksisia rakeita, mikä johtaa kohdesolun hajoamiseen.
MHC I:n ja NK-soluja estävien reseptorien geenit vaihtelevat ihmisten välillä. Näiden geenien alleelit assosioituvat mm. joidenkin autoimmuunisairauksien riskiin. NK-solut muuttuvat iän myötä, ja niihin vaikuttavat krooniset virusinfektiot, kuten sytomegalovirus (CMV). Koska
NK-solut kykenevät tappamaan kasvainsoluja, ne ovat syöpähotoihin kohdistuvan immuuniterapian kannalta kiinnostava tutkimuskohde. Jotkut terapeuttiset monoklonaaliset vasta -aineet perustuvat NK-solujen hyödyntämiseen. Tutkijat kehittävät myös hoitoja NK -solujen aktivoimiseksi käyttämällä sytokiinejä ja testaavat geneettisesti muunnettuja eläviä NK-soluja immuuniterapiana.
Luonnolliset tappajasolut ja immuunijärjestelmä
Luontaisen (innate) immuunijärjestelmän luonnollisia tappajasoluja ei tule sekoittaa hankinnaisen (adaptive) immuunijärjestelmn luonnollisiin tappaja T-soluihin (Natural killer T cell).
Luonnolliset tappajasolut, jotka tunnetaan myös nimellä NK-solut tai suuret granulaariset lymfosyytit (LGL) ovat synnynnäiseen/luontaiseen (innate) immuunijärjestelmään kuuluvia sytotoksisia (eli infektoituneita soluja tappavia) lymfosyyttejä. NK-solut kuuluvat kasvavaan synnynnäisten lymfoidisolujen (ILC) perheeseen, jotka muodostavat 5–20 prosenttia kaikista elimistön lymfosyyteistä [1].
NK-solujen rooli on verrannollinen hankinnaisen (adaptive) immuunivasteen sytotoksisten T-solujen roolin kanssa.
Luontaisen immuniteetin järjestelmät torjuvat nopeasti ja tehokkaasti mikrobeja ja muita elimistöön päässeitä tai kertyneitä vieraita aineksia tilanteissa, joissa antigeenejä spesifisesti tunnistavia vasta-aineita ja sytotoksisia T-soluja ei ole ehtinyt muodostua, sekä tilanteissa, joissa adaptiivinen immuniteetti tarvitsee tehostemekanismeja tunnistamiensa vieraiden rakenteiden siivoamiseen. NK-solut aktivoituvat nopeasti (noin kolmessa vuorokaudessa) viruksen tai jonkin muun patogeenin aiheuttamaan infektioon.
MHC (Major Histocompatibility Complex)
Tavallisesti immuunisolut havaitsevat MHC*-glykoproteiinien muutokset infektion saaneiden solujen pinnoilla. MHC-proteiineja kutsutaan usein myös HLA*-antigeeneiksi. MHC-molekyylejä on kahta tyyppiä: luokan I MHC- ja luokan II MHC-molekyylit.
Luokan I MHC-proteiineja ilmennetään lähes kaikissa selkärankaisen solutyypeissä. Ne esittelevät antigeenejä sytotoksisille T-soluille. Luokan II MHC-proteiineja ilmennetään pääasiassa soluissa, jotka ovat vuorovaikutuksessa auttaja-T-solujen (T helper-cell) kanssa, kuten dendriittisoluissa, makrofageissa ja B-lymfosyyteissä.
MHC-proteiineja esiintyy sairastuneiden solujen solupinnoilla, mikä laukaisee sytokiinien erittymisen. Tämä johtaa infektoidun solun lyyttiseen hajoamiseen (lysis) tai apoptoosiin (ohjattu solukuolema).
NK-soluja kutsutaan ”luonnollisiksi tappajiksi”, koska niiden immuunivaste ei edellytä erityistä aktivointia ja solun infektioon viittaava MHC I-markkeria [2]. Tämän vuoksi NK-solujen immuunireaktio on nopea. Muut immuunisolut, kuten T-solut, eivät havaitse ja tuhoa haitallisia soluja, joista puuttuu nämä MHC I -markkerit.
MHC-proteiinit toimivat immunologisessa puolustuksessa sitoen antigeeninä toimivan proteiinin pilkkoutumisen seurauksena syntyneitä peptidifragmentteja ja kuljettaen niitä antigeeniä esittelevän solun (dendriittisolu naiivien T-solujen tapauksessa) pinnalle, jossa ne esitellään yhdessä muiden stimulatoristen signaalien kanssa T-soluille.
Tämän seurauksena aktivoituneet efektori-T-solut tunnistavat saman peptidi-MHC-kompleksin kohdesolunsa pinnalla, mikä voi sytotoksisten T-solujen tapauksessa olla mikä tahansa infektoitunut elimistön solu, auttaja-T-solujen tapauksessa B-solu, sytotoksinen T-solu, infektoitunut makrofagi tai dendriittisolu itse.
* major histocompatibility complex
* human leucocyte-associated antigens

schematic diagram indicating the complementary activies of cytotoxic T cells and NK cells
NK-solut tehostavat synnynnäisen immuunijärjestelmän toimintaa
Luonnolliset tappajasolut kuuluvat synnynnäisten imusolujen ryhmään. Ne ovat yksi kolmesta solutyypistä, jotka ovat eriytyneet tavallisista imukudoksen kantasoluista. Ryhmän kaksi muuta imusolutyyppiä ovat B- ja T -solut. NK-solut erottaa muista lymfosyyteistä CD56:n ilmentyminen ja CD3:n puuttuminen (CD56+, CD3−) [3, 4].
NK -solut erilaistuvat ja kypsyvät luuytimessä, imusolmukkeissa, pernassa, risoissa ja kateenkorvassa [5]. Luonnolliset tappajasolut eroavat luonnollisista tappaja-T-soluista (NKT) fenotyypin, alkuperän ja efektoritoimintojen perusteella. Usein NKT-soluaktiivisuus edistää NK-solujen toimintaa erittämällä gamma-interferonia.
NK-solut eivät ilmennä T-soluantigeenireseptoreita (TCR), pan-T-merkkiaineita (CD3), tai pinta-immunoglobuliinien (Ig) B-solureseptoreita, mutta yleensä ne ilmentävät pintamarkkereita, kuten CD16 ja CD57. NKp46 -solun pintamarkkeri on NK -solumarkkeri, joka ilmentyy ihmisisen solujen lisäksi monien eläinten soluissa [6] [7].
Luonnolliset tappajasolut tehostavat luontaisen immunijärjestelmän toimintaa. Aktivoivilla ja inhiboivilla NK -solureseptoreilla on tärkeitä toiminnallisia tehtäviä immunologisen toleranssin (self tolerance) ja NK -solujen aktiivisuuden ylläpitämisessä.
NK-solut osallistuvat myös adaptiiviseen immuunivasteeseen [8]. Kokeet ovat osoittaneet NK-solujen kyvyn mukautua välittömään ympäristöön ja muodostaa antigeenispesifistä immunologista muistia, joka on olennainen sekundaarisiin infektioihin vastaamisessa samalla antigeenillä [9].
NK -solujen rooli immuunivasteissa on tärkeä tutkimuslinja tutkimuksessa, jossa käytetään NK -solujen aktiivisuutta mahdollisena syöpähoitona.
* MeSH = Medical Subject Headings
* FMA = The Foundational Model of Anatomy Ontology
NK-solujen historia
Varhaisissa syöpäpotilaiden tutkimuksissa soluvälitteisestä sytotoksisuudesta kasvainkohdesoluja vastaan havaittiin johdonmukaisesti niin sanottua ”luonnollista” reaktiivisuutta. Jotkin lymfosyytti-populaatiot hajottivat poikkeuksellisesti kasvainsoluja ilman, että niitä oli aikaisemmin herkistetty kasvainsoluille.
Tohtori Henry Smith (Leedsin yliopiston lääketieteellinen korkeakoulu) julkaisi havainnosta tutkimuksen (1966), jossa vahvistettiin, että käsittelemättömät lymfoidisolut pystyivät antamaan luonnollisen immuniteetin kasvaimille [10]. Monet tutkijat olivat tehneet vastaavia havaintoja, mutta koska löydöt olivat ristiriidassa tuolloin vakiintuneen mallin kanssa, monet pitivät näitä havaintoja artefakteina*.[11]
| * Artefakti (lääketiede): 1. keinotekoinen tuote; rakenne tai piirre, joka ei ole luonnollinen, vaan johtuu manipulaatiosta. 2. kuvan vääristymä tai sumeus, joka johtuu käsittelystä tai varastoinnista. |
Vuoteen 1973 mennessä ”luonnollinen tappava” reaktiivisuus vahvistettiin useilla lajeilla. Havaintojen seurauksena postuloitiin oletus erillisen solulinjan olemassaolosta, jolla on tämä kyky. Havainnon, että ainutlaatuinen lymfosyyttityyppi oli vastuussa spontaanista sytotoksisuudesta, teki 1970-luvun alussa Rolf Kiessling ja Hugh Pross hiirillä [12] sekä Hugh Pross ja Mikael Jondal ihmisillä [13][14]. Hiiri- ja ihmistutkimukset toteutettiin Tukholman Karoliinisen instituutin professorien Eva Kleinin ja Hans Wigzellin ohjauksessa.
Kiesslingin tutkimus koski T-lymfosyyttien hyvin karakterisoitua kykyä hajottaa kasvainsoluja, joita vastaan ne oli aiemmin immunisoitu. Pross ja Jondal tutkivat soluvälitteistä sytotoksisuutta ihmisen veressä ja erilaisten reseptoria sisältävien solujen poistamisen vaikutusta tähän sytotoksisuuteen. Myöhemmin samana vuonna Ronald Herberman julkaisi samanlaisia havaintoja hiiren efektorisolun ainutlaatuisesta luonteesta [15]. Ihmisillä tehtyjen tutkimusten tiedot vahvisti suurimmaksi osaksi West et al.[16] käyttäen samanlaisia tekniikoita ja samaa erytroleukeemista kohdesolulinjaa, K562. K562 on herkkä ihmisen NK-solujen hajoamiselle. Sittemmin K562 51-kromin vapautumismäärityksestä on tullut yleisimmin käytetty määritys ihmisen NK-toiminnallisen aktiivisuuden havaitsemiseksi [17].
Menetelmän yleinen käyttö on johtanut siihen, että laboratoriot ympäri maailmaa voivat helposti vertailla kokeellisia tietoja. Käyttämällä epäjatkuvaa tiheyssentrifugointia ja monoklonaalisia vasta-aineita, luonnollinen tappamiskyky kartoitettiin suurten, granulaaristen lymfosyyttien alaryhmään (LGL), jotka tunnetaan nykyään NK-soluina.
Timonen ja Saksela osoittivat vuonna 1980, että tiheysgradientilla eristetyt suuret granulaariset lymfosyytit olivat vastuussa ihmisen NK-aktiivisuudesta [18]. Tämä oli ensimmäinen kerta, kun NK-solut visualisoitiin mikroskooppisesti, ja se oli suuri läpimurto alalla.
NK-solujen alaryhmät
NK-solut luokitellaan:
CD56bright NK-solut erittävät sytokiineja ja ovat toiminnaltaan samankaltaisia kuin T-auttajasolut [20]. CD56bright NK-solut muodostavat valtaosan luuytimen, toissijaisista imukudoselimistä, maksan ja iho NK-soluista [3].
| Imukudosta on myös imukeräsissä. Imukeräsiä löytyy risakudoksista, Peyerin levyistä suolistossa, umpilisäkkeestä ja hengitysteiden limakalvolta. Näistä imukudoksista käytetään nimitystä MALT-järjestelmä (mucosa-associated lymphoid tissue). Imukudoselimet voidaan jakaa ensisijaisiin imukudoselimiin, joita ovat luuydin ja kateenkorva sekä toissijaisiin imukudoselimiin, joita ovat perna, risat, imusuonisto imusolmukkeineen, erilliset imukeräset ja imukerässikermät. |
CD56dim NK-soluja löydetään pääasiassa perifeerisestä verestä*, [3]. Näitä NK-soluja luonnehtii ”kyky tappaa” sairastuneita soluja [20]. CD56dim NK-solut ovat aina CD16-positiivisia (CD16 on vasta-aine-riippuvaisen soluvälitteinen sytotoksisuus (ADCC – Antibody-dependent cellular cytotoxicity)) välittäjä [20]. CD56bright NK-solut voivat muuttua CD56dim -soluiksi CD16 välityksellä [3]. Luonnolliset tappajasolut tuhoavat viruksen infektoimia soluja CD16-välitteisesti vasta-aine-riippuvaisessa soluvälitteisessä sytotoksisuudessa (ADCC)[21].
Kaikilla COVID-19-potilailla CD56bright NK-solujen toiminta on heikentynyt, mutta CD56dim on heikentynyt vain vakavaa COVID-19-infektiota sairastavilla [21].
| * Perifeerinen veri kulkee sydämen, valtimoiden, kapillaarien ja verisuonien läpi. Sen tärkein tehtävä on kuljettaa happea ja ravintoaineita elimistön soluihin ja kudoksiin sekä poistaa hiilidioksidia ja muita kuona-aineita elimistöstä. |
NK-solujen reseptorit
NK-solureseptorit voidaan erottaa toiminnan perusteella. Luonnolliset sytotoksisuusreseptorit indusoivat suoraan apoptoosin (solukuoleman) Fas-ligandiin sitoutumisen jälkeen, mikä osoittaa suoraan solun tartunnan.
MHC-riippumattomat reseptorit käyttävät vaihtoehtoista reittiä apoptoosin indusoimiseksi infektoiduissa soluissa. Luonnollinen tappajasoluaktivaatio määräytyy estävän ja aktivoivan reseptoristimulaation tasapainon perusteella. Esimerkiksi, jos inhiboiva reseptorin signalointi on näkyvämpää, NK-soluaktiivisuus estyy; samoin, jos aktivoiva signaali on hallitseva, seurauksena on NK-solujen aktivaatio [22]. NK-solureseptorityypit (joissa on inhiboivia ja joitain aktivoivia jäseniä) erotetaan rakenteesta. Seuraavassa on muutama esimerkki:
Aktivoivat reseptorit
- Ly49 (homodimeeri, eli kahdesta rakenneyksiköstä koostuva rakenne, jonka molemmat yksiköt ovat samanlaisia). Ihmisellä on yksi pseudogeeninen Ly49-reseptori, joka kuuluu vanhoihin C-tyypin lektiiniperheen reseptoreihin. Ly49 on klassisten (polymorfisten) MHC I -molekyylien reseptori.
- NCR (luonnolliset sytotoksisuusreseptorit), immunoglobuliinien superperheen tyypin 1 transmembraaniproteiinit välittävät stimulaatiossa NK:n tappamista ja IFNy:n vapautumista. Ne sitovat virusligandeja, kuten hemagglutiniinit ja hemagglutiniinineuraminidaasit, joitain bakteeriligandeja ja soluligandeja, jotka liittyvät kasvaimen kasvuun, kuten PCNA.
- CD16 (FcγIIIA) CD16 on erilaistumismolekyylien ryhmä, joka löytyy luonnollisten tappajasolujen, neutrofiilien, monosyyttien ja makrofagien pinnasta. Se osallistuu vasta-aineriippuvaiseen soluvälitteiseen sytotoksisuuteen.CD16 on immunoglobuliinien superperheen (IgSF) molekyyli, joka osallistuu vasta-aineriippuvaiseen solusytotoksisuuteen (ADCC). Sitä voidaan käyttää spesifisten immuunisolujen populaatioiden eristämiseen fluoresenssiaktivoidun solulajittelun (FACS) tai magneettisesti aktivoidun solulajittelun avulla käyttämällä CD16:ta vastaan suunnattuja vasta-aineita.CD16 on tyypin III Fcy-reseptori. Ihmisillä sitä esiintyy kahdessa eri muodossa: FcyRIIIa (CD16a) ja FcyRIIIb (CD16b), joiden sekvenssien samankaltaisuus on 96 % solunulkoisilla immunoglobuliinia sitovilla alueilla. Kun FcyRIIIa ekspressoituu syöttösoluissa, makrofageissa ja luonnollisissa tappajasoluissa transmembraanisena reseptorina, FcyRIIIb ekspressoituu vain neutrofiileissä.
FcyRIIIb on ainoa Fc-reseptori, joka on ankkuroitu solukalvoon glykosyylifosfatidyyli-inositoli (GPI) -linkkerillä, ja sillä on myös merkittävä rooli kalsiumin mobilisaation ja neutrofiilien degranulaation käynnistämisessä. FcγRIIIa ja FcγRIIIb pystyvät yhdessä aktivoimaan degranulaation, fagosytoosin ja oksidatiivisen purskeen, mikä mahdollistaa neutrofiilien puhdistamisen opsonisoituneista patogeeneista.
Inhiboivat (estävät) reseptorit
- Tappajasolun immunoglobuliininkaltaiset reseptorit (KIRs) kuuluvat Ig:n kaltaisten solun ulkoisten domeenien reseptorien multigeeniseen perheeseen. Ne ovat klassisen MHC I:n (HLA-A, HLA-B, HLA-C) sekä ei-klassisen Mamu-G:n (HLA-G) pääreseptoreita kädellisillä. Jotkut KIR:t ovat spesifisiä tietyille HLA-alatyypeille. Useimmat KIR:t ovat estäviä ja hallitsevia. Tavalliset solut ilmentävät MHC-luokkaa I, joten KIR-reseptorit tunnistavat ne ja NK-solujen sytotoksinen reaktio estyy.
- CD94/NKG2 (heterodimeeri, eli kahdesta rakenneyksiköstä koostuva rakenne, jonka molemmat yksiköt ovat rakenteeltaan erilaisia) on C-tyypin lektiiniperheen reseptori, joka on konservoitunut (säilynyt samanlaisena) sekä jyrsijöillä että kädellisillä ja identifioi ei-klassisia (myös ei-polymorfisia) MHC I -molekyylejä, kuten HLA-E. HLA-E:n ilmentyminen solun pinnalla riippuu klassisten MHC-luokan I molekyylien signaalisekvenssistä peräisin olevan nonameeripeptidiepitoopin läsnäolosta, joka muodostuu signaalipeptidipeptidaasin ja proteasomin peräkkäisestä toiminnasta.
- ILT tai LIR (immunoglobuliininkaltainen reseptori) kuuluu äskettäin tunnistettuun Ig-reseptoreiden perheeseen.
- Ly49:llä on sekä aktivoivia että estäviä isoformeja. Ne ovat erittäin polymorfisia populaatiotasolla; vaikka ne eivät ole rakenteellisesti sukua KIR:eille, ne ovat KIR:ien toiminnallisia homologeja hiirillä. Ly49:t ovat klassisten (polymorfisten) MHC I -molekyylien reseptoreita.
Funktio
Sytolyyttinen granulaatiovälitteinen soluapoptoosi
NK-solut ovat sytotoksisia (solulle myrkyllisiä).Pienet granulat sytoplasmassa sisältävät proteiineja, kuten perforiinia ja proteaaseja, eli grantsyymeja. Vapautuessaan tapettavaksi tarkoitetun solun välittömään läheisyyteen perforiini muodostaa huokosia kohdesolun solukalvoon, luoden näin vesipitoisen kanavan, jonka kautta grantsyymit ja niihin liittyvät molekyylit pääsevät soluun, mikä indusoi joko apoptoosin tai lyyttiseen solun hajoamisen.
Ero apoptoosin ja solun hajoamisen välillä on tärkeä immunologiassa: viruksen saastuttaman solun hajottaminen voi mahdollisesti vapauttaa virioneja, kun taas apoptoosi johtaa viruksen tuhoutumiseen. NK-solut erittävät myös α-defensiinejä, antimikrobisia molekyylejä, ja ne tappavat bakteereja suoraan hajottamalla niiden soluseiniä neutrofiilien kanssa analogisella tavalla.[5]
Vasta-aineriippuvainen soluvälitteinen sytotoksisuus (ADCC)
Infektoituneet solut opsonoidaan rutiininomaisesti vasta-aineilla immuunisolujen havaitsemiseksi. NK-soluissa ilmentyvät FcyRIII (CD16) -reseptorit voivat tunnistaa antigeeneihin sitoutuvat vasta-aineet, mikä johtaa NK-aktivaatioon, sytolyyttisten rakeiden vapautumiseen ja siitä johtuvaan soluapoptoosiin. Tämä on joidenkin monoklonaalisten vasta-aineiden, kuten rituksimabin (Rituxan), ofatumumabin (Azera) ja muiden, tärkeä toimintamekanismi. Vasta-aineriippuvaisen soluvälitteisen sytotoksisuuden vaikutus kasvainsolujen tappamiseen voidaan mitata spesifisellä testillä, joka käyttää NK-92:ta, kuolematonta NK-kaltaisten solujen linjaa, joka on lisensoitu NantKwest, Inc:lle: NK-92-solujen vaste, joka on transfektoitu korkean affiniteetin Fc-reseptorilla verrataan ”villin tyypin” NK-92:een, joka ei ilmennä Fc-reseptoria.[23]
Sytokiinien indusoima NK-solujen ja sytotoksisten T-lymfosyyttien (CTL) aktivaatio
Sytokiineilla on ratkaiseva rooli NK-solujen aktivoinnissa. Koska nämä ovat stressimolekyylejä, joita solut vapauttavat virusinfektion seurauksena, ne toimivat signaalina NK-soluille viruspatogeenien esiintymisestä sairastuneella alueella.
NK-aktivaatioon osallistuvia sytokiinejä ovat IL-12, IL-15, IL-18, IL-2 ja CCL5. NK-solut aktivoituvat vasteena interferoneille tai makrofageista peräisin oleville sytokiineille. Ne toimivat virusinfektioiden hillitsemisessä, kun taas adaptiivinen immuunivaste tuottaa antigeenispesifisiä sytotoksisia T-soluja, jotka voivat poistaa infektion.
NK-solut hallitsevat virusinfektioita erittämällä IFNy:aa ja TNFa:aa. IFNy aktivoi makrofageja fagosytoosia ja hajoamista varten, ja TNFa toimii edistäen suoraa NK-kasvainsolujen tappamista. Potilaat, joilta puuttuu NK-soluja, ovat erittäin alttiita herpesvirusinfektion varhaisille vaiheille.
Puuttuva itsensä tunnistamisen -hypoteesi
Jotta NK-solut voisivat puolustaa kehoa viruksia ja muita taudinaiheuttajia vastaan, ne tarvitsevat mekanismeja, joilla ne tunnistavat onko solu infektoitunut vai ei. Tarkat mekanismit ovat edelleen tutkimuksen kohteena, mutta ”muuttuneen minän” tilan tunnistamisen uskotaan liittyvän asiaan.
Sytotoksisen aktiivisuutensa hallitsemiseksi NK-soluilla on kahden tyyppisiä pintareseptoreita: aktivoivat reseptorit ja estävät reseptorit sekä tappajasolujen immunoglobuliinin kaltaiset reseptorit. Useimmat näistä reseptoreista eivät ole ainutlaatuisia NK-soluille, ja niitä voi esiintyä myös joissakin T-solujen alaryhmissä.
Estävät reseptorit tunnistavat MHC-luokan I alleelit, mikä selittää, miksi NK-solut ensisijaisesti tappavat soluja, joissa on vähän MHC-luokan I molekyylejä. Tämä NK-solukohteen vuorovaikutusmuoto tunnetaan nimellä ”puuttuva itsensä tunnistaminen”. Klas Kärre kollegoineen nimesi hypoteesin 90-luvun lopulla.
MHC-luokan I molekyylit ovat päämekanismi, jolla solut näyttävät virus- tai kasvainantigeenejä sytotoksisille T-soluille. Sekä solunsisäisissä mikrobeissa että kasvaimissa on havaittavissa yleinen evoluutionaalinen sopeutuminen: MHC I -molekyylien krooninen vaimeneminen, mikä tekee sairastuneista soluista näkymättömiä T-soluille, jolloin ne voivat välttää T-soluvälitteisen immuunivasteen. NK-solut ilmeisesti kehittyivät evoluutioreaktiona tähän sopeutumiseen (MHC:n menetys eliminoi CD4/CD8-toiminnan, joten toinen immuunisolu kehittyi täyttämään tehtävän)[24].
Kasvainsolujen seuranta
Luonnollisista tappajasoluista puuttuu usein antigeenispesifisiä solupintareseptoreita, joten ne ovat osa synnynnäistä (luontaista) immuunijärjestelmää, eli ne pystyvät reagoimaan välittömästi ilman aiempaa altistusta taudinaiheuttajalle.
Sekä hiirillä että ihmisillä NK-soluilla voidaan nähdä rooli kasvaimen immuunivalvonnassa indusoimalla suoraan kasvainsolujen kuolemaa (NK:t toimivat sytolyyttisinä efektorilymfosyytteinä), jopa ilman pintaadheesiomolekyylejä ja antigeenisiä peptidejä. Tämä NK-solujen rooli on tärkeä immuunijärjestelmän toiminnalle erityisesti siksi, että T-solut eivät pysty tunnistamaan patogeenejä pinta-antigeenien puuttuessa [2].
Kasvainsolujen havaitseminen johtaa NK-solujen aktivoitumiseen ja siitä johtuvaan sytokiinien tuotantoon ja erittymiseen. Jos kasvainsolut eivät aiheuta tulehdusta, niitä pidetään myös itsenä, eivätkä ne aiheuta T-soluvastetta.
NK:t tuottavat useita sytokiinejä, mukaan lukien tuumorinekroositekijä a (TNFa), IFNy ja interleukiini (IL-10). TNFa ja IL-10 toimivat proinflammatorisina ja vastaavasti immunosuppressoreina.
NK-solujen aktivaatio ja sitä seuraava sytolyyttisten efektorisolujen tuotanto vaikuttaa makrofageihin, dendriittisoluihin ja neutrofiileihin, mikä mahdollistaa sen jälkeen antigeenispesifiset T- ja B-soluvasteet. Sen sijaan, että se vaikuttaisi antigeenispesifisten reseptorien kautta, NK-solujen aiheuttamaa kasvainsolujen hajoamista välittävät vaihtoehtoiset reseptorit, mukaan lukien NKG2D, NKp44, NKp46, NKp30 ja DNAM [22]. NKG2D on disulfidisidottu homodimeeri, joka tunnistaa useita ligandeja, mukaan lukien ULBP ja MICA, jotka tyypillisesti ilmentyvät kasvainsoluissa. Dendriittisolujen ja NK-solujen välisen rajapinnan roolia immunobiologiassa on tutkittu ja määritelty kriittiseksi monimutkaisen immuunijärjestelmän ymmärtämiselle.
NK-solut, makrofagit ja eräät muut solutyypit, ilmentävät Fc-reseptori-molekyyliä (FcR) – (FC-gamma-RIII = CD16). Se on aktivoiva biokemiallinen reseptori, joka sitoo IgG-luokan vasta-aineiden Fc-osan. Tämä sallii NK-solujen kohdistaa aktivaationsa soluihin ja hajottaa soluja vasta-aineriippuvaisen soluvälitteisen sytotoksisuuden (ADCC) kautta.
Immuunivaste riippuu NK-soluissa ilmennetyn Fc-reseptorin affiniteetista, jolla voi olla korkea, keskitasoinen ja alhainen affiniteetti vasta-aineen Fc-osaan. Tämän affiniteetin määrää proteiinin asemassa 158 oleva aminohappo, joka voi olla fenyylialaniini (F-alleeli) tai valiini (V-alleeli). Potilaat, joilla on korkea affiniteetti FcRgammRIII (158 V/V alleeli), reagoivat paremmin vasta-ainehoitoon. Tämä on osoitettu lymfoomapotilailla, jotka ovat saaneet Rituxan-vasta-ainetta.
Potilailla, jotka ilmentävät 158 V/V alleelia, oli parempi antituumorivaste. Vain 15–25 % väestöstä ilmentää 158 V/V alleelia. Monoklonaalisten vasta-aineiden ADCC-osuuden määrittämiseksi NK-92-solut (”puhdas” NK-solulinja) on transfektoitu korkean affiniteetin FcR:n geenillä.
Vanhenevien solujen poistaminen
Luonnollisilla tappajasoluilla ja makrofageilla on tärkeä tehtävä vanhenevien solujen poistamisessa [25]. Luonnolliset tappajasolut tappavat vanhenevia soluja ja erittävät makrofageja aktivoivia sytokiinejä, jotka aktivoivat makrofagit poistamaan vanhenevia soluja [25].
Luonnolliset tappajasolut voivat käyttää NKG2D-reseptoreita havaitsemaan vanhenevia soluja ja tappamaan nämä solut käyttämällä perforiinihuokosia muodostavaa sytolyyttistä proteiinia.[26] CD8+ sytotoksiset T-lymfosyytit käyttävät myös NKG2D-reseptoreita havaitsemaan vanhenevia soluja ja edistämään NK-solujen tapaan vanhenevien solujen tappamista.[26]
NK-solujen adaptiiviset ominaisuudet – ”muistin kaltaiset”, ”mukautuvat” ja muisti-NK-solut
Kyky synnyttää muistisoluja primaarisen infektion jälkeen sekä siitä seuraava nopea immuunivaste saman antigeenin aiheuttamiin myöhempiin infektioihin on keskeinen osa T- ja B-solujen tehtäviä adaptiivisessa immuunivasteessa.
NK-soluja on pidetty osana luontaista immuunijärjestelmää. Viime aikoina lisääntynyt näyttö kuitenkin viittaa siihen, että NK-soluilla voi olla useita ominaisuuksia, jotka yleensä johtuvat adaptiivisista immuunisoluista (esim. T-soluvasteista), kuten alaryhmien dynaaminen laajeneminen ja supistuminen, lisääntynyt pitkäikäisyys ja immunologisen muistin muoto, jolle on ominaista tehokkaampi vaste saman antigeenin toissijaiseen altistukseen [27][28].
Hiirien MCMV-mallissa havaittiin MCMV-indusoitujen NK-solujen suojaavia muistitoimintoja [29] ja MCMV-ligandin m157 suora tunnistaminen Ly49-reseptorin toimesta osoittautui tärkeäksi adaptiivisten NK-soluvasteiden synnyttämisessä.[29]
Ihmisillä useimmat tutkimukset ovat keskittyneet aktivoivaa reseptoria NKG2C (KLRC2) kantavan NK-solujen alajoukon laajentamiseen. Tällaista laajenemista havaittiin ensisijaisesti vasteena ihmisen sytomegalovirukselle (HCMV) [30], mutta myös muissa infektioissa, mukaan lukien hantavirus, Chikungunya-virus, HIV ja virushepatiitti.
Laukaisevatko virusinfektiot adaptiivisten NKG2C+ NK-solujen laajentumisen vai johtavatko muut infektiot piilevän HCMV:n uudelleen aktivoitumiseen (kuten hepatiitista on arveltu [31]), on vielä ratkaisematta. Viimeaikaiset tutkimukset viittaavat siihen, että adaptiiviset NK-solut voivat käyttää aktivoivaa reseptoria NKG2C (KLRC2) sitoutuakseen suoraan sytomegaloviruksesta peräisin oleviin peptidiantigeeneihin ja reagoida peptidien tunnistamiseen aktivoimalla, laajentamalla ja erilaistumalla [32]. Tämä mekanismi, joka reagoi viruksen infektoimiin soluihin tunnettiin aiemmin vain adaptiivisen immuunijärjestelmän T-solujen immuunivasteesta.
NK-solujen toiminta raskausaikana
Koska siittiöiden mukana naisen munasoluun päätyy vieraita geenejä, onnistunut hedelmöittyminen edellyttää äidin immuunijärjestelmän tukahduttamista. NK-soluilla uskotaan olevan keskeinen merkitys tässä prosessissa [33]. Kohdun NK-solut (uNK-solut) eroavat perifeerisistä NK-soluista.
Kohdun NK-solut kuuluvat CD56bright NK -solujen alaryhmään. Ne erittävät sytokiineja, mutta niillä on alhainen sytotoksinen kyky. Ne ovat verrattain samankaltaisia perifeeristen CD56bright NK-solujen kanssa, mutta niillä on hieman erilainen reseptoriprofiili.[33]
uNK-solut ovat runsaimpia leukosyyttejä kohdussa raskauden alkuvaiheessa. Niiden osuus on noin 70 % kohdun leukosyyteistä, mutta niiden alkuperästä ei ole varmuutta [34]. Kohdun NK-solut tuottavat solusytotoksisuutta in vitro, mutta vähemmän kuin perifeeriset NK-solut, vaikka ne sisältävät perforiinia [35].
Sytotoksisuuden puute in vivo voi johtua kohdun NK-solujen estoreseptoreiden ligandeista. Trofoblastisolut vähentävät HLA-A:n ja HLA-B:n säätelyä suojautuakseen sytotokselta T-soluvälitteiseltä kuolemalta. Tämä aktivoisi normaalisti NK-solut puuttuvalla itsetunnistuksella; nämä solut kuitenkin säilyvät. Trofoblastin HLA-E:n (joka on ligandi NK-soluja inhiboivalle reseptorille NKG2A) ja HLA-G:n (joka on ligandi NK-soluja estävälle reseptorille KIR2DL4) selektiivisen retention uskotaan puolustavan sitä NK-soluvälitteistä kuolemaa vastaan. [33]
Perifeeristen NK-solujen korkeampia prosenttiosuuksia esiintyy naisilla, joilla on ollut toistuvia keskenmenoja [36].
NK-solut ovat vuorovaikutuksessa HLA-C:n kanssa tuottaen sytokiinejä, jotka ovat välttämättömiä trofoblastiselle proliferaatiolle. Joitakin tärkeitä sytokiineja, joita ne erittävät, ovat TNF-α, IL-10, IFN-y, GM-CSF ja TGF-β [33]. Esimerkiksi IFN-y laajentaa ja ohentaa äidin kierrevaltimoiden seinämiä parantaakseen verenkiertoa implantaatiokohtaan [37].
Syöpäsolut ja NK-solut
Tuumorisolut voivat välttää immuunivasteet poistamalla NKG2D:n liukoisia ligandeja. Nämä liukoiset NKG2D-ligandit sitoutuvat NK-solujen NKG2D-reseptoreihin aktivoiden väärän NK-vasteen ja siten luoden kilpailua reseptoripaikasta [2]. Tätä kiertotapaa esiintyy eturauhassyövässä. Lisäksi eturauhassyövän kasvaimet voivat välttää CD8-solujen tunnistamisen, koska ne pystyvät vaimentamaan MHC-luokan I molekyylien ilmentymistä. Tämä esimerkki immuunijärjestelmän väistämisestä korostaa itse asiassa NK-solujen merkitystä kasvaimen seurannassa ja vasteessa, koska CD8-solut voivat näin ollen vaikuttaa vain kasvainsoluihin vasteena NK:n käynnistämään sytokiinien tuotantoon (adaptiivinen immuunivaste) [38].
NK-solujen korkea pitoisuus
Kokeelliset hoidot NK-soluilla ovat johtaneet liialliseen sytokiinituotantoon ja jopa septiseen sokkiin. Tulehduksellisen sytokiini gamma-interferonin ehtyminen käänsi vaikutuksen.
Sovellukset
Syöpäterapiat
Koska NK-solut tunnistavat kohdesolut, kun ne ilmentävät ei-omia HLA-antigeenejä (mutta eivät itseään), autologiset (potilaiden omat) NK-soluinfuusiot eivät ole osoittaneet kasvainten vastaisia vaikutuksia.
Sen sijaan tutkijat työskentelevät perifeerisen veren allogeenisten solujen käyttämiseksi, mikä edellyttää, että kaikki T-solut poistetaan ennen infuusiota, jotta voidaan poistaa käänteishyljintäsairauden riski, joka voi olla kohtalokas.
Tämä voidaan saavuttaa käyttämällä immunomagneettista kolonnia (CliniMACS). Lisäksi, koska veressä on rajoitettu määrä NK-soluja (vain 10 % lymfosyyteistä on NK-soluja), niiden määrää on lisättävä viljelmässä. Tämä voi kestää muutaman viikon ja tuotto riippuu luovuttajasta.
Yksinkertaisempi tapa saada suuria määriä puhtaita NK-soluja on laajentaa NK-92-soluja, joiden solut kasvavat jatkuvasti viljelmässä ja jotka voidaan laajentaa kliinisen asteen määrään pusseissa tai bioreaktoreissa [39]. Kliiniset tutkimukset ovat osoittaneet sen olevan hyvin siedetty, ja joitakin kasvaimia estäviä vasteita on havaittu potilailla, joilla on keuhkosyöpä, melanooma ja lymfooma [40][41].
NK-92-immunoterapiaan liittyy kuitenkin merkittäviä rajoituksia, koska solulinja on peräisin potilaalta, jolla on non-Hodgkin-lymfooma, ja siksi se on säteilytettävä ennen infuusiota, mikä rajoittaa pysyvyyttä in vivo. Lisäksi NK-92-soluista puuttuu CD-16, minkä vuoksi ne eivät pysty suorittamaan ADCC:tä, mikä estää tämän hoidon käyttämisen yhdessä monoklonaalisten vasta-ainehoitojen kanssa [42]. Ne voidaan kuitenkin suunnitella sisältämään CD16, mikä mahdollistaa ADCC-toiminnan ja laajentaa niiden mahdollista terapeuttista käyttökelpoisuutta.
T-solujen infuusiot, jotka on suunniteltu ilmentämään kimeeristä antigeenireseptoria (CAR), joka tunnistaa antigeenimolekyylin leukemiasoluissa, voivat saada aikaan remissioita potilailla, joilla on pitkälle edennyt leukemia. T-solujen laajenemiseen liittyy logistisia haasteita, ja tutkijat työskentelevät soveltaakseen samaa tekniikkaa ääreisveren NK-soluihin ja NK-92:een. NK-92-soluja voidaan muokata sisältämään sekä CD16- että CAR-soluja, jotta ne voivat suorittaa sekä ADCC-välitteisen tappamisen IgG1-vasta-aineiden kautta että CAR-välitteisen tappamisen samasta solusta. Yksi tällainen NK-92:sta johdettu solulinja, nimeltään t-haNK, on muokattu sekä CD16:n että anti-PD-L1 CAR:n kanssa, ja se on parhaillaan kliinisessä kehityksessä onkologisiin indikaatioihin. NK-92.
Bostonin lastensairaalassa tehdyssä tutkimuksessa, yhteistyössä Dana-Farber Cancer Instituten kanssa, jossa immuunipuutteiset hiiret olivat saaneet lymfoomia EBV-infektiosta, NK-aktivoiva reseptori NKG2D, fuusioitiin EBV-vasta-aineen stimuloivan Fc-osan kanssa. NKG2D-Fc-fuusio vähensi kasvaimen kasvua ja pidensi vastaanottajien elinaikaa. LMP1-käyttöisten lymfoomien siirtomallissa NKG2D-Fc-fuusio vähensi kasvaimen kasvua ja pidensi vastaanottajien elinaikaa. Hodgkin-lymfoomassa, jossa pahanlaatuiset Hodgkin Reed-Sternberg -solut ovat tyypillisesti HLA-luokan I puutteellisia, immuunijärjestelmän väistäminen välittyy osittain vinoutumisesta kohti uupunutta PD-1hi NK -solufenotyyppiä, ja näiden NK-solujen uudelleenaktivoituminen näyttää olevan yksi tarkistuspisteen estämisen aiheuttama vaikutusmekanismi.[43]
Uudet havainnot
Luontainen resistenssi HIV:lle
Tuoreet tutkimukset viittaavat siihen, että spesifiset KIR-MHC luokan I geenivuorovaikutukset voivat hallita synnynnäistä geneettistä vastustuskykyä tietyille virusinfektioille, mukaan lukien HIV [5]. Tiettyjen HLA-allotyyppien on havaittu määrittävän HIV:n etenemisen AIDSiksi; esimerkkinä ovat HLA-B57- ja HLA-B27-alleelit, joiden on havaittu hidastavan etenemistä HIV:stä AIDSiin. Tämä on ilmeistä, koska potilailla, jotka ilmentävät näitä HLA-alleeleja, on havaittu olevan pienempi viruskuorma ja CD4+ T-solujen lukumäärän asteittainen lasku.
Huolimatta huomattavasta HLA-alleelien ja KIR-allotyyppien geneettistä korrelaatiota mittaavasta tutkimuksesta ja kerätyistä tiedoista, varmaa johtopäätöstä siitä, mikä yhdistelmä vähentää HIV- ja AIDS-alttiutta, ei ole vielä tehty. NK-solut voivat aiheuttaa immuunipaineen HIV:lle, mikä on aiemmin kuvattu vain T-soluilla ja vasta-aineilla [44]. HIV mutatoituu välttääkseen NK-solujen havaitsemisen [44].
Kudosten NK-solut
Suurin osa nykyisestä tiedostamme on peräisin hiiren pernan ja ihmisen perifeerisen veren NK-solujen tutkimuksista.
Viime vuosina on kuvattu kudoksissa eläviä NK-solupopulaatioita [45][46]. Nämä kudoksessa elävät NK-solut jakavat transkription samankaltaisuuden kuin aiemmin kuvatut kudoksessa asuvat muisti-T-solut. Kuitenkaan kudoksissa asuvat NK-solut eivät välttämättä ole muistifenotyyppiä, ja itse asiassa suurin osa kudoksessa olevista NK-soluista on toiminnallisesti epäkypsiä[47].
Näillä erikoistuneilla NK-solujen alajoukoilla voi olla rooli elinten homeostaasissa. Esimerkiksi NK-solut rikastuvat ihmisen maksassa tietyllä fenotyypillä ja osallistuvat maksafibroosin hallintaan [48][49]. Kudoksissa eläviä NK-soluja on myös tunnistettu sellaisista kohdista kuin luuytimessä, pernassa ja viime aikoina keuhkoissa, suolistossa ja imusolmukkeissa. Näissä paikoissa kudoksissa asuvat NK-solut voivat toimia säiliönä kypsymättömien NK-solujen ylläpitämiselle ihmisissä läpi elämän.[47]
Lähde: Wikipedia, Imperial College London
Lähdeluettelo
- Perera Molligoda Arachchige, Arosh Shavinda (2021-03-24). ”Human NK cells: From development to effector functions”. Innate Immunity. 27 (3): 212–229. doi:10.1177/17534259211001512. ISSN 1753-4259. PMC 8054151. PMID 33761782.
- Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S (January 2011). ”Innate or adaptive immunity? The example of natural killer cells”. Science. 331 (6013): 44–9. Bibcode:2011Sci…331…44V. doi:10.1126/science.1198687. PMC 3089969. PMID 21212348.
- Pfefferle A, Jacobs B, Sohlberg E, Malmberg K (2020). ”Deciphering Natural Killer Cell Homeostasis”. Frontiers in Immunology. 11: 812. doi:10.3389/fimmu.2020.00812. PMC 7235169. PMID 32477340.
- Roitt I, Brostoff J, Male D (2001). Immunology (6th ed.), 480p. St. Louis: Mosby, ISBN 0-7234-3189-2.
- Iannello A, Debbeche O, Samarani S, Ahmad A (July 2008). ”Antiviral NK cell responses in HIV infection: I. NK cell receptor genes as determinants of HIV resistance and progression to AIDS”. Journal of Leukocyte Biology. 84 (1): 1–26. CiteSeerX 10.1.1.619.9639. doi:10.1189/jlb.0907650. PMID 18388298. S2CID 26975415.
- Walzer T, Bléry M, Chaix J, Fuseri N, Chasson L, Robbins SH, Jaeger S, André P, Gauthier L, Daniel L, Chemin K, Morel Y, Dalod M, Imbert J, Pierres M, Moretta A, Romagné F, Vivier E (February 2007). ”Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46”. Proceedings of the National Academy of Sciences of the United States of America. 104 (9): 3384–9. Bibcode:2007PNAS..104.3384W. doi:10.1073/pnas.0609692104. PMC 1805551. PMID 17360655.
- Sivori S, Vitale M, Morelli L, Sanseverino L, Augugliaro R, Bottino C, Moretta L, Moretta A (October 1997). ”p46, a novel natural killer cell-specific surface molecule that mediates cell activation”. The Journal of Experimental Medicine. 186 (7): 1129–36. doi:10.1084/jem.186.7.1129. PMC 2211712. PMID 9314561.
- Arina A, Murillo O, Dubrot J, Azpilikueta A, Alfaro C, Pérez-Gracia JL, Bendandi M, Palencia B, Hervás-Stubbs S, Melero I (May 2007). ”Cellular liaisons of natural killer lymphocytes in immunology and immunotherapy of cancer”. Expert Opinion on Biological Therapy. 7 (5): 599–615. doi:10.1517/14712598.7.5.599. PMID 17477799. S2CID 43003664.
- Watzl C (2014). How to trigger a killer: modulation of natural killer cell reactivity on many levels. Advances in Immunology. 124. pp. 137–70. doi:10.1016/B978-0-12-800147-9.00005-4. ISBN 9780128001479. PMID 25175775.
- Smith HJ (December 1966). ”Antigenicity of carcinogen-induced and spontaneous tumours in inbred mice”. British Journal of Cancer. 20 (4): 831–7. doi:10.1038/bjc.1966.95. PMC 2008147. PMID 5964614.
- Oldham RK (1983). ”Natural killer cells: artifact to reality: an odyssey in biology”. Cancer and Metastasis Reviews. 2 (4): 323–36. doi:10.1007/BF00048565. PMID 6375859. S2CID 11301147.
- Kiessling R, Klein E, Pross H, Wigzell H (February 1975). ””Natural” killer cells in the mouse. II. Cytotoxic cells with specificity for mouse Moloney leukemia cells. Characteristics of the killer cell”. European Journal of Immunology. 5 (2): 117–21. doi:10.1002/eji.1830050209. PMID 1086218. S2CID 2389610.
- Pross HF, Jondal M (August 1975). ”Cytotoxic lymphocytes from normal donors. A functional marker of human non-T lymphocytes”. Clinical and Experimental Immunology. 21 (2): 226–35. PMC 1538269. PMID 810282.
- Jondal M, Pross H (April 1975). ”Surface markers on human b and t lymphocytes. VI. Cytotoxicity against cell lines as a functional marker for lymphocyte subpopulations”. International Journal of Cancer. 15 (4): 596–605. doi:10.1002/ijc.2910150409. PMID 806545. S2CID 30612835.
- Herberman RB, Nunn ME, Holden HT, Lavrin DH (August 1975). ”Natural cytotoxic reactivity of mouse lymphoid cells against syngeneic and allogeneic tumors. II. Characterization of effector cells”. International Journal of Cancer. 16 (2): 230–9. doi:10.1002/ijc.2910160205. PMID 1080480. S2CID 24410880.
- West WH, Cannon GB, Kay HD, Bonnard GD, Herberman RB (January 1977). ”Natural cytotoxic reactivity of human lymphocytes against a myeloid cell line: characterization of effector cells”. Journal of Immunology. 118 (1): 355–61. PMID 299761.
- Pross HF, Baines MG, Rubin P, Shragge P, Patterson MS (January 1981). ”Spontaneous human lymphocyte-mediated cytotoxicity against tumor target cells. IX. The quantitation of natural killer cell activity”. Journal of Clinical Immunology. 1 (1): 51–63. doi:10.1007/BF00915477. PMID 7334070. S2CID 24437710.
- Timonen T, Saksela E (1980). ”Isolation of human NK cells by density gradient centrifugation”. Journal of Immunological Methods. 36 (3–4): 285–91. doi:10.1016/0022-1759(80)90133-7. PMID 7430655.
- Hashemi E, Malarkannan S (2020). ”Tissue-Resident NK Cells: Development, Maturation, and Clinical Relevance”. Cancers. 12 (6): 1553. doi:10.3390/cancers12061553. PMC 7352973. PMID 32545516.
- Wu S, Fu T, Jiang Y, Shao Z (2020). ”Natural killer cells in cancer biology and therapy”. Molecular Cancer. 19 (1): 120. doi:10.1186/s12943-020-01238-x. PMC 7409673. PMID 32762681.
- Market M, Angka L, Martel AB, Auer RC (2020). ”Flattening the COVID-19 Curve With Natural Killer Cell Based Immunotherapies”. Frontiers in Immunology. 11: 1512. doi:10.3389/fimmu.2020.01512. PMC 7324763. PMID 32655581.
- Terunuma H, Deng X, Dewan Z, Fujimoto S, Yamamoto N (2008). ”Potential role of NK cells in the induction of immune responses: implications for NK cell-based immunotherapy for cancers and viral infections”. International Reviews of Immunology. 27 (3): 93–110. doi:10.1080/08830180801911743. PMID 18437601. S2CID 27557213.
- Smyth MJ, Hayakawa Y, Takeda K, Yagita H (November 2002). ”New aspects of natural-killer-cell surveillance and therapy of cancer”. Nature Reviews. Cancer. 2 (11): 850–61. doi:10.1038/nrc928. PMID 12415255. S2CID 1430364.
- Lodoen MB, Lanier LL (2005). ”Viral modulation of NK cell immunity”. Nature Reviews Microbiology. 3 (1): 59–69. doi:10.1038/nrmicro1066. PMID 15608700. S2CID 16655783.
- Antonangeli F, Zingoni A, Soriani A, Santoni A (2019). ”Senescent cells: Living or dying is a matter of NK cells”. Journal of Leukocyte Biology. 105 (6): 1275–1283. doi:10.1002/JLB.MR0718-299R. PMID 30811627. S2CID 73469394.
- Prata LG, Ovsyannikova IG, Tchkonia T, Kirkland JL (2018). ”Senescent cell clearance by the immune system: Emerging therapeutic opportunities”. Seminars in Immunology. 40: 101275. doi:10.1016/j.smim.2019.04.003. PMC 7061456. PMID 31088710.
- Rölle A, Pollmann J, Cerwenka A (September 2013). ”Memory of infections: an emerging role for natural killer cells”. PLOS Pathogens. 9 (9): e1003548. doi:10.1371/journal.ppat.1003548. PMC 3784484. PMID 24086127.
- Pyzik M, Vidal SM (2009). ”Natural killer cells: NK cells stroll down the memory lane”. Immunology and Cell Biology. 87 (4): 261–3. doi:10.1038/icb.2009.10. PMID 19290015. S2CID 42943696.
- Sun JC, Beilke JN, Lanier LL (January 2009). ”Adaptive immune features of natural killer cells”. Nature. 457 (7229): 557–61. Bibcode:2009Natur.457..557S. doi:10.1038/nature07665. PMC 2674434. PMID 19136945.
- Gumá M, Angulo A, Vilches C, Gómez-Lozano N, Malats N, López-Botet M (December 2004). ”Imprint of human cytomegalovirus infection on the NK cell receptor repertoire”. Blood. 104 (12): 3664–71. doi:10.1182/blood-2004-05-2058. PMID 15304389.
- Malone DF, Lunemann S, Hengst J, Ljunggren HG, Manns MP, Sandberg JK, Cornberg M, Wedemeyer H, Björkström NK (2017). ”Cytomegalovirus-Driven Adaptive-Like Natural Killer Cell Expansions Are Unaffected by Concurrent Chronic Hepatitis Virus Infections”. Frontiers in Immunology. 8 (8): 525. doi:10.3389/fimmu.2017.00525. PMC 5421146. PMID 28533779.
- Hammer Q, Rückert T, Borst EM, Dunst J, Haubner A, Durek P, Heinrich F, Gasparoni G, Babic M, Tomic A, Pietra G, Nienen M, Blau IW, Hofmann J, Na IK, Prinz I, Koenecke C, Hemmati P, Babel N, Arnold R, Walter J, Thurley K, Mashreghi MF, Messerle M, Romagnani C (May 2018). ”Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells”. Nature Immunology. 19 (5): 453–463. doi:10.1038/s41590-018-0082-6. PMID 29632329. S2CID 4718187.
- Lash GE, Robson SC, Bulmer JN (March 2010). ”Review: Functional role of uterine natural killer (uNK) cells in human early pregnancy decidua”. Placenta. 31 Suppl (S): S87–92. doi:10.1016/j.placenta.2009.12.022. PMID 20061017.
- Bulmer JN, Williams PJ, Lash GE (2010). ”Immune cells in the placental bed”. The International Journal of Developmental Biology. 54 (2–3): 281–94. doi:10.1387/ijdb.082763jb. PMID 19876837.
- Kopcow HD, Allan DS, Chen X, Rybalov B, Andzelm MM, Ge B, Strominger JL (October 2005). ”Human decidual NK cells form immature activating synapses and are not cytotoxic”. Proceedings of the National Academy of Sciences of the United States of America. 102 (43): 15563–8. Bibcode:2005PNAS..10215563K. doi:10.1073/pnas.0507835102. PMC 1266146. PMID 16230631.
- Seshadri S, Sunkara SK (2013). ”Natural killer cells in female infertility and recurrent miscarriage: a systematic review and meta-analysis”. Human Reproduction Update. 20 (3): 429–38. doi:10.1093/humupd/dmt056. PMID 24285824.
- Ashkar AA, Di Santo JP, Croy BA (July 2000). ”Interferon gamma contributes to initiation of uterine vascular modification, decidual integrity, and uterine natural killer cell maturation during normal murine pregnancy”. The Journal of Experimental Medicine. 192 (2): 259–70. doi:10.1084/jem.192.2.259. PMC 2193246. PMID 10899912.
- O’Leary JG, Goodarzi M, Drayton DL, von Andrian UH (May 2006). ”T cell- and B cell-independent adaptive immunity mediated by natural killer cells”. Nature Immunology. 7 (5): 507–16. doi:10.1038/ni1332. PMID 16617337. S2CID 1459858.
- Gong JH, Maki G, Klingemann HG (April 1994). ”Characterization of a human cell line (NK-92) with phenotypical and functional characteristics of activated natural killer cells”. Leukemia. 8 (4): 652–8. PMID 8152260.
- Arai S, Meagher R, Swearingen M, Myint H, Rich E, Martinson J, Klingemann H (2008). ”Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial”. Cytotherapy. 10 (6): 625–32. doi:10.1080/14653240802301872. PMID 18836917.
- Tonn T, Becker S, Esser R, Schwabe D, Seifried E (August 2001). ”Cellular immunotherapy of malignancies using the clonal natural killer cell line NK-92”. Journal of Hematotherapy & Stem Cell Research. 10 (4): 535–44. doi:10.1089/15258160152509145. PMID 11522236.
- Matosevic, S (2018). ”Viral and Nonviral Engineering of Natural Killer Cells as Emerging Adoptive Cancer Immunotherapies”. J Immunol Res. 2018: 4054815. doi:10.1155/2018/4054815. PMC 6166361. PMID 30306093.
- Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, Cui Q, Han E, Tobin J, Bird R, Cross D, Hernandez A, Gould C, Birch S, Gandhi MK (April 2018). ”Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages Is More Prominent in Hodgkin Lymphoma Than DLBCL”. Blood. 131 (16): 1809–1819. doi:10.1182/blood-2017-07-796342. PMC 5922274. PMID 29449276.
- Alter G, Heckerman D, Schneidewind A, Fadda L, Kadie CM, Carlson JM, Oniangue-Ndza C, Martin M, Li B, Khakoo SI, Carrington M, Allen TM, Altfeld M (August 2011). ”HIV-1 adaptation to NK-cell-mediated immune pressure”. Nature. 476 (7358): 96–100. doi:10.1038/nature10237. PMC 3194000. PMID 21814282.
- Yokoyama WM, Sojka DK, Peng H, Tian Z (2013-01-01). ”Tissue-resident natural killer cells”. Cold Spring Harbor Symposia on Quantitative Biology. 78: 149–56. doi:10.1101/sqb.2013.78.020354. PMID 24584057.
- Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, Zhong C, Chase JM, Rothman PB, Yu J, Riley JK, Zhu J, Tian Z, Yokoyama WM (January 2014). ”Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells”. eLife. 3: e01659. doi:10.7554/elife.01659. PMC 3975579. PMID 24714492.
- Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, Kubota M, Matsumoto R, Thapa P, Szabo PA, Li Poon MM, Li J, Arakawa-Hoyt J, Shen Y, Fong L, Lanier LL, Farber DL (February 2020). ”Tissue Determinants of Human NK Cell Development, Function, and Residence”. Cell. 180 (4): 749–763. e13. doi:10.1016/j.cell.2020.01.022. PMC 7194029. PMID 32059780.
- Hudspeth K, Donadon M, Cimino M, Pontarini E, Tentorio P, Preti M, Hong M, Bertoletti A, Bicciato S, Invernizzi P, Lugli E, Torzilli G, Gershwin ME, Mavilio D (January 2016). ”Human liver-resident CD56(bright)/CD16(neg) NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways”. Journal of Autoimmunity. 66: 40–50. doi:10.1016/j.jaut.2015.08.011. PMC 4718768. PMID 26330348.
- Fasbender F, Widera A, Hengstler JG, Watzl C (2016). ”Natural Killer Cells and Liver Fibrosis”. Frontiers in Immunology. 7: 19. doi:10.3389/fimmu.2016.00019. PMC 4731511. PMID 26858722.
Luettavaa:
- Perera Molligoda Arachchige A. S. (2021). Human NK cells: From development to effector functions. Innate immunity, 17534259211001512. Advance online publication. https://doi.org/10.1177%2F17534259211001512
- Cellular and Molecular Immunology by Abul K. Abbas & Andrew Lichtman Saunders Copyright 2003
- How the Immune System Works, 2nd edition, by Lauren Sompayrac, PhD Blackwell Publishing 2003
- Immunobiology: The Immune System In Health And Disease by Janeway, Travers, Walport & Shlomchik Churchchill Livingstone Copyright 2005
- Kuby Immunology, 6th edition, by Thomas J. Kindt, Richard A. Goldsby, and Barbara A. Osborne, W.H. Freeman and Company, New York