Ruokavalio ja vanheneminen: molekyylibiologinen näkökulma
Samo Ribarič1
1Institute of Pathophysiology, Medical Faculty, University of Ljubljana, Zaloška 4, 1000 Ljubljana, Slovenia
Tiivistelmä
Ravitsemuksella on merkittäviä ja pitkäaikaisia terveysvaikutuksia, jotka eivät rajoitu vain yksilöön, vaan voivat siirtyä yksilöltä seuraavalle sukupolvelle. Se voi myötävaikuttaa kroonisten sairauksien kehittymiseen ja etenemiseen, mikä edelleen vaikuttaa odotettavissa olevaan elinikään.
Ruokavaliolla voi pidentää odotettavissa olevaa elinikää ja parantaa yleistä terveyttä. Tässä artikkelissa selitetään biokemialliset mekanismit, joihin tällainen rohkea väite perustuu. Artikkeli on hyvin haastava. Yleissääntönä on, että ravinnosta saadun energian rajoittaminen, paasto, pätkäpaasto ja paastoa aineenvaihdunnan tasolla imitoiva ketogeeninen ruokavalio aktivoivat kehossa solutason mekanismeja, jotka ylläpitävät solujen hyvinvointia ja pitkäikäisyyttä.
Kalorirajoitus (CR) voi pidentää keskimääräistä elinikää ja viivästyttää ikään liittyvien muutosten alkamista monissa organismeissa. Energian rajoittaminen saa aikaan koordinoituja adaptiivisia stressivasteita solujen ja koko organismin tasolla moduloimalla epigeneettisiä mekanismeja (esim. DNA:n metylaatio, transtrationaaliset histonimodifikaatiot), signaalireittejä, jotka säätelevät solujen kasvua ja ikääntymistä (esim. TOR, AMPK, p53 ja FOXO) ja solusta soluun signalointimolekyylejä (esim. adiponektiini).
Näiden mukautuvien stressivasteiden kokonaisvaikutuksena on lisääntynyt vastustuskyky stressille, mikä viivästyttää ikään liittyviä fysiologisia muutoksia ja edistää pitkäikäisyyttä. Kalorirajoitus voi hillitä monia ikääntymiseen liittyviä sairauksia, kuten syöpiä, diabetesta, ateroskleroosia, sydän- ja verisuonitauteja sekä hermostoa rappeuttavia sairauksia.
Vaihtoehtona kaloreiden rajoittamiselle on tutkittu useita kaloreita rajoittavia ruokavalioita eläimillä ja ihmisillä. Tällä hetkellä lupaavimmat vaihtoehdot kalorirajoituksen käytölle ihmisillä näyttävät olevan liikunnan lisääminen yksin tai yhdessä vähentyneen kalorien saannin kanssa.
Samo Ribarič’in laaja artikkeli tarkastelee ruokavalion merkitystä vanhenemiseen aineenvaihdunnan ja biokemian perspektiivistä. Monet tässä esiin nostetut asiat hyödyttävät kaikkia.
Tämä ei ole aivan helppolukuinen artikkeli. Liitän tekstiin aihetta käsitteleviä videoita, jotka helpottavat erilaisten aineenvaihduntapolkujen, ylävirran tapahtumien ja alasreguloivien modulaattorien maailman kartoittamista.
Samo Ribaričin artikkeli julkaistiin 2012. Sen jälkeen tieto ketogeenisen ruokavalion, paaston ja pätkäpaaston hyödyistä on lisääntynyt. Vaikka artikkeli vain sivuaa ohimennen niitä, se sisältää erinomaisia selityksiä vanhenemiseen ja terveyteen vaikuttavista molekyylibiologian mekanismeista, jotka nykytietämyksen mukaan toteutuvat myös KD-ruokavaliossa, pätkäpaastossa ja paastossa.
1. Johdanto
Ravitsemuksella on merkittäviä pitkäaikaisia vaikutuksia terveyteen. Se on sellainen elämäntapaan liittyvä tekijä, joka voi edistää tai vähentää kroonisten sairauksien, kuten sydän- ja verisuonitautien, diabeteksen ja syövän riskiä [1].
Kroonisten sairauksien ehkäisyn ja hallinnan pitäisi olla globaali prioriteetti, koska krooniset sairaudet aiheuttavat yli puolet kaikista kuolemantapauksista [2]. Sairastuminen on potilaille ja potilaiden omaisille henkisesti raskas taakka. Kroonisten sairauksien hoitokulut rasittavat yhteiskunnan kantokykyä. Kuolemaa ei kukaan voi välttää, mutta terveisiin elinvuosiin jokainen voi vaikuttaa omilla elämäntapavalinnoillaan.
Ravitsemuksen vaikutukset terveyteen eivät rajoitu yksilöön, vaan ne voivat siirtyä yksilöltä seuraavalle sukupolvelle. Tämä havainto on vahvistettu epidemiologisilla tutkimuksilla ja eläinkokeilla.
Pienenä syntyvän vauvan riski sairastua myöhemmin sepelvaltimotautiin, tyypin 2 diabetekseen ja lihavuuteen on normaalipainoisina syntyviä lapsia selvästi korkeampi [3–7]. Eläinmallissa synnytystä edeltävä aliravitsemus laski jälkeläisten elinikää [8] tai johti nefronien puutteelliseen kehitykseen, mikä lisäsi kroonisen munuaissairauden riskiä myöhemmässä elämässä [9]. (Nefroni on munuaisen toiminnallinen yksikkö, joka suodattaa virtsaa verestä ja säätelee virtsan määrää sekä koostumusta.)
2. Ruokavaliotekijöiden epigeneettiset muutokset
Ravitsemuksen vaikutukset kehoon välittyvät epigeneettisillä mekanismeilla [1]. McKay’n ja Mathersin mukaaan kolme tunnettua läheisesti vuorovaikutuksessa olevaa mekanismia ovat DNA:n metylaatio, histonimodifikaatio ja koodaamattomat mikroRNA:t (miRNA:t) [1].
Artikkelin toinen luku sisältää melkoisesti molekyylibiologian jargonia. Hyppää kolmanteen lukuun suoraan, jos tämä vaikuttaa tylsältä.
Ravintotekijät voivat indusoida epigeneettisiä muutoksia kolmen reitin kautta: (a) suora vaikutus geenien ilmentymiseen, (b) tumareseptorien aktivaatio ligandien avulla ja (c) membraanireseptorien (solukalvoreseptorien) signalointikaskadien muokkaus [10].
Epigeneettiset mekanismit tarjoavat organismeille tehokkaan aikaan reagoivan järjestelmän geeniekspression mukauttamiseksi:
(a) kudostyyppispesifisesti
(b) organismin kehitystilaan sopivasti
(c) kehon ulkoisen ja sisäisen ympäristön signaalit huomioiden [1].
2.1. DNA-metylaatio ruokavaliolla
Folaatti ja B12-vitamiini edistävät laajaa DNA-metylaatiota, kun taas seleeni, vihreän teen polyfenolit ja bioflavonoidit vähentävät yleistä DNA-metylaatiota, Davis et al. [17].
DNA-metylaatio on kudosspesifinen ja sitä säätelee DNA-metyylitransferaasi (DNMT) -entsyymi, joka modifioi sytosiiniemäksen CpG-dinukleotiditähteessä metyyliryhmän kanssa muodostaen 5-metyylisytosiinin [11].
Esimerkkejä DNA-metyloinnilla kontrolloiduista prosesseista ovat X-kromosomien inaktivaatio, iturataspesifisten geenien leimaaminen ja hiljentäminen, karsinogeneesi ja pitkäaikaisen muistin muodostuminen [12].
Perinteisesti DNA-metylaatio on liitetty geeniekspression tukahduttamiseen. Siten DNA-metylaatio joko fyysisesti estää transkriptioproteiinien sitoutumista geeniin, tai metyloitu DNA sitoutuu proteiineihin, jotka tunnetaan metyyli-CpG:tä sitovina domeeniproteiineina. Ne rekrytoivat ylimääräisiä proteiineja lokukseen – kuten histonideasetylaasit – jotka muuttavat histoneja kompaktiksi inaktiiviseksi kromatiiniksi, kuten on osoitettu [13, 14].
Joillakin syöpäpotilailla esiintyy sekä laajaa DNA-hypometylaatiota että paikallista DNA-hypermetylaatiota [15, 16]. Ruokavalion ainesosia, joiden tiedetään moduloivan DNA-metylaatiota, ovat esimerkiksi folaatti, B12–vitamiini, seleeni, vihreän teen polyfenolit (esim. epigallokatekiini-3-gallaatti (EGCG), epikatekiini, ganisteiini) ja bioflavonoidit (kvertsetiini, fisetiini ja myrisetiini).
Folaatti ja B12-vitamiini edistävät laajaa DNA-metylaatiota, kun taas seleeni, vihreän teen polyfenolit ja bioflavonoidit vähentävät yleistä DNA-metylaatiota, Davis et al. [17]. Näiden aineosien paikallinen vaikutus DNA-metylaatioon voi kuitenkin poiketa niiden laajasta vaikutuksesta. Esimerkiksi seleenin pitkäaikainen kulutus lisää p53-geenin eksonispesifistä DNA-metylaatiota rotan maksassa ja paksusuolen limakalvossa [18].
2.2. Histonien modifiointi ruokavaliolla
Aitotumallisten eukaryoottien solujen tumat sisältävät tiiviisti pakattuna emäksisiä proteiineja (johtuen positiivisesti varautuneesta N-päästä, jossa on monia lysiini- ja arginiinitähteitä), joita kutsutaan histoneiksi.
Histonit pakkaavat ja järjestävät DNA:n rakenneyksiköiksi, eli nukleosomeiksi.
Kromatiinin perusyksikön, nukleosomin ytimen muodostavat 2 kpl kutakin histonia H2A, H2B, H3 ja H4 eli yht. 8 molekyyliä (ns. oktameerirakenne). Histonimolekyylit muodostavat litteän kiekon tai kelan, jossa DNA muodostaa 2 kierrosta histonien ympäri ja näin täydentää tuman rakenteen.
Histonit toimivat keloina, joiden ympärillä DNA pyörii ja joilla on rooli geenien säätelyssä. Aktiiviset geenit ovat vähemmän sitoutuneita histoneihin; inaktiiviset geenit liittyvät voimakkaasti histoneihin [19].
Histonien N-terminaali (histonihäntä) tai sivuketjut pallomaisessa histonisydämessä ovat epigeneettisten modifikaatioiden kohdat [20]. Histonien translaation jälkeinen modifikaatio on merkittävästi monimuotoisempi kuin DNA:n metylaatio. Jotkut parhaiten ymmärretyistä histonimodifikaatioista ovat metylointi, asetylointi, fosforylaatio, ribosylointi, ubikitinointi, sumoylaatio tai biotinylointi [20].
Esimerkkejä histonien translaation jälkeiseen modifikaatioon osallistuvista entsyymeistä ovat histoniasetyylitransferaasit (HAT), metyylitransferaasit (HMT), deasetylaasit (HDAC) ja demetylaasit (HDM).
Ruokavalion vaikutuksia histonin translaation jälkeiseen modifikaatioon tarkastelivat hiljattain mm. Link et al. [21]. Esimerkiksi valkosipulin ja kanelin polyfenolit estävät HDAC:ia; vihreän teen polyfenolit ja kupari estävät HAT:ia; EGCG estää HMT:ia.
Histonin metylaatio voi moduloida DNA:n metylaatiokuvioita, ja DNA:n metylaatio voi toimia mallina joillekin histonimuutoksille DNA:n replikaation jälkeen [20, 22]. On arveltu, että nämä vuorovaikutukset voitaisiin toteuttaa suorilla vuorovaikutuksilla histonin ja DNA-metyylitransferaasien välillä [20, 22]. Tällaiset DNA-histoni-vuorovaikutukset voidaan moduloida myös ruokavalion avulla.
2.3. miRNA-modulointi ruokavaliolla
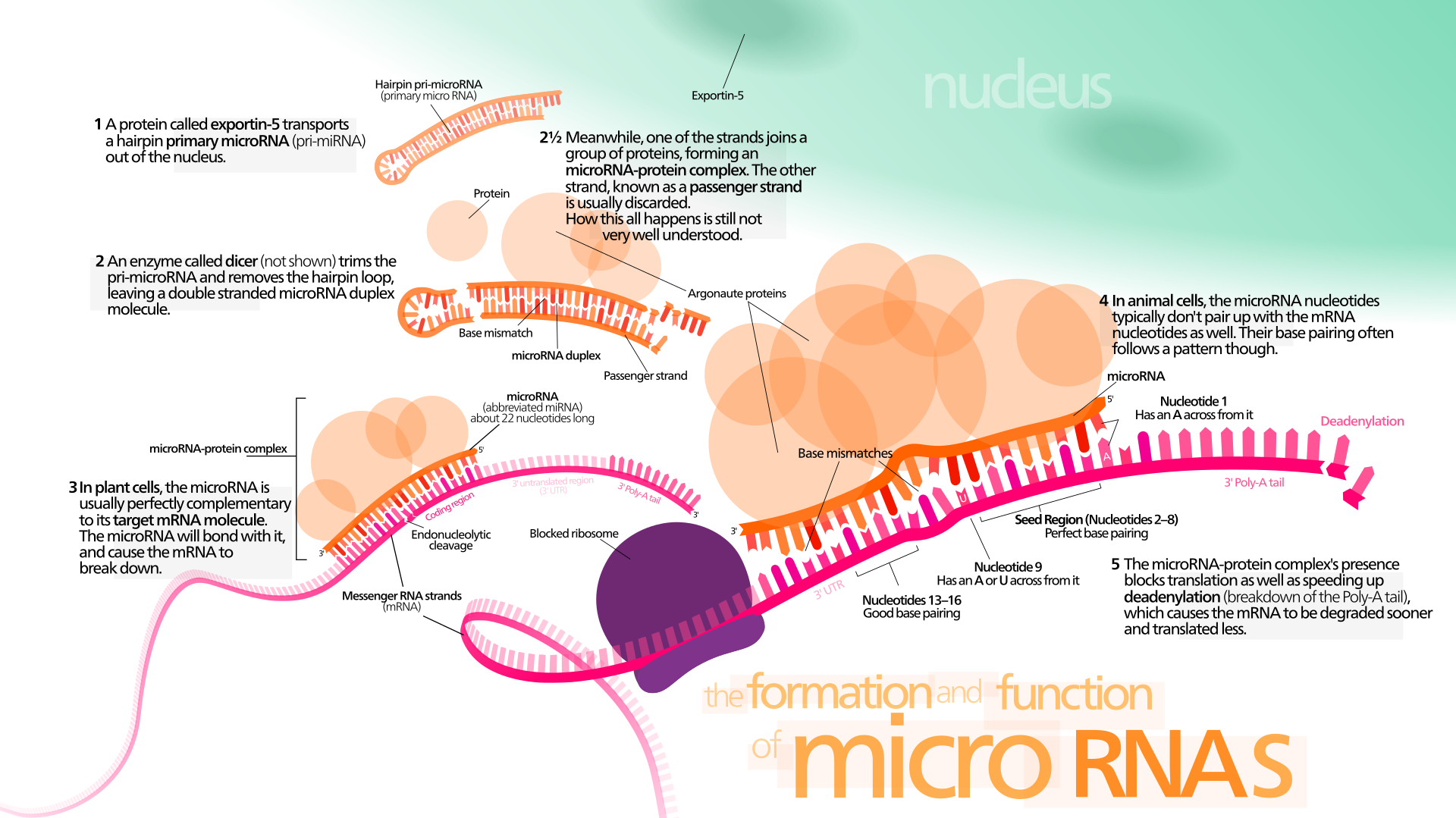
Kaikilla elävillä soluilla on kyky vastaanottaa ja käsitellä solukalvojen ulkopuolelta tulevia signaaleja.
Eukaryoottien eli aitotumallisten miRNA (mikro-RNA) on lyhyt, parin kymmenen nukleotidin pituinen, yksijuosteinen RNA-molekyyli, joka estää tietyn lähetti-RNA:n toiminnan kiinnittymällä siihen eli se hiljentää geenin. Monet mikro-RNA:ista ovat proteiinia koodaamattomilta DNA-alueilta.
MikroRNA (miRNA) on ei-koodaava RNA-molekyyli (joka sisältää 22 nukleotidia). miRNA toimii RNA:n hiljentämisessä ja geeniekspression jälkitranskriptiossa. Niitä löytyy kasveista, eläimistä ja joistakin viruksista.
miRNA:t toimivat emäspariliitoksen kautta mRNA-molekyylien komplementaaristen sekvenssien kanssa. Tämän seurauksena nämä mRNA-molekyylit hiljennetään yhdellä tai useammalla seuraavista menetelmistä: (1) mRNA-juosteen pilkkominen kahteen osaan, (2) mRNA:n destabilisointi lyhentämällä sen poly (A) häntää ja ( 3) Vähemmän tehokas mRNA:n translaatio proteiineiksi ribosomien avulla.
miRNA:t ovat transkriptionaalisia säätelijöitä ja sitoutuvat komplementaarisiin sekvensseihin kohde-lähetti-RNA-transkripteissa (mRNA:t), mikä johtaa transkriptionaalisten geenien hiljentymiseen mRNA-translaation repressoinnin tai lisääntyneen RNA-hajoamisen vuoksi.
miRNA:t voivat kuitenkin myös aiheuttaa histonimodifikaatiota ja promoottorikohtien DNA-metylaatiota, mikä säätelee kohdegeenien ilmentymistä vaihtoehtoisella reitillä. [23, 24]. Ihmisen genomi koodaa yli 1000 miRNA-nukleotidia, joiden kohteena on 50% nisäkäsgeeneistä monissa ihmisen solutyypeissä [25–30].
Siten miRNA:t vaikuttavat monien transkriptiotekijöiden, reseptorien ja kuljettajien ilmentymiseen [31]. Viimeaikaiset havainnot ihmis- ja eläinmalleissa tehdyistä kokeista viittaavat siihen, että ravitsemus (esim. rasvan, proteiinin, alkoholin tai E-vitamiinin kulutus) vaikuttaa monien miRNA-nukleotidien [32] ilmentymiseen.
miRNA:t muistuttavat RNA-interferenssi (RNAi) -reitin pieniä häiritseviä RNA:ita (siRNA:t), paitsi että miRNA:t ovat peräisin RNA-transkriptioiden alueista, kun taas siRNA:t ovat peräisin pitkistä kaksijuosteisen RNA:n alueista. Ihmisen genomi voi koodata yli 1900 miRNA:a, vaikka uudempi analyysi osoittaa, että luku on lähempänä 600: ta. Kiertävät miRNA:t vapautuvat kehon nesteisiin; vereen ja aivo-selkäydinnesteeseen. Ne toimivat biomarkkereina monissa sairauksissa.
Monet miRNA:t ovat evoluutiokonservoituneita, mikä tarkoittaa, että niillä on tärkeät biologiset toiminnot, joilla ei ole suuria lajienvälisiä eroja. Esimerkiksi 90 miRNA-perhettä on säilynyt ainakin nisäkkäiden ja kalojen yhteisestä esi-isästä lähtien, ja suurimmalla osalla näistä konservoiduista miRNA:ista on tärkeitä tehtäviä.
Polyfenolit (esim. antosyaniini, kurkumiini ja kvertsetiini) moduloivat maksan miRNA:n ilmentymistä in vivo hiirimalleissa [33]. miRNA:n ilmentymisen modulointi ruokavaliolla voi selittää genisteiinin, kurkumiinin, retinoiinihapon ja kalaöljyn syövältä suojaavia vaikutuksia.
Genisteiini (isoflavoni) estää uveaalisen melanoomasolun kasvua estämällä miRNA-27a:n ilmentymistä [34]. Kurkumiinihoito säätelee miRNA-22:n ja alasreguloidun miRNA-199a:n ilmentymistä haimasyöpäsolulinjassa [35] ja säätelee myös miRNA-15a:n ja miRNA-16:n ilmentymistä rintasyöpäsoluissa [36].
Akuuttia promyelosyyttistä leukemiaa sairastavilla potilailla, joita hoidettiin menestyksekkäästi kemoterapialla ja all-trans-retinoiinihapolla, miRNA-181b:n säätely alasreguloitui (downregulate), mutta monien muiden miRNA:iden säätely ylösreguloitiin (upregulate) [37]*.
Retinoiinihappohoidon indusoima miRNA-10a-säätely esti haimasyövän etäpesäkkeitä ksenotransplantaatiokokeissa seeprakalan alkioissa [38]. Kalaöljy vähensi erilaisten ekspressoitujen miRNA:iden määrää koe-eläimissä ja voi olla hyödyllistä paksusuolikarsinooman estämisessä [39]. Indol-3-karbinoli sääteli useiden miRNA:iden (ts. miRNA:iden -21, -31, -130a, -146b ja -377) ilmentymistä hiirissä, joille oli indusoitu hiiren keuhkokasvaimia [40].
Ravintoaineiden puutos voi myös moduloida miRNA:n ilmentymistä. Esimerkiksi folaatin puute liittyi miRNA-222:n merkittävään yli-ilmentymiseen [41]. Myös rotilla, joilla oli folaatti-metioniini-koliini-puutteellinen ruokavalio, kehittyi maksasolujen karsinooma, johon liittyi samanaikaisesti miRNA:iden yli-ilmentymistä -17 – -92, -21, -23, -130 ja -190 [42].
*Ylös- ja alasregulaatio
Alasregulaatio tarkoittaa prosessia, jossa jokin solun ulkoinen ärsyke vähentää RNA:n tai proteiinin määrää, kun taas ylösregulaatio tai sääntelyn lisääminen lisää näitä komponentteja solussa.
Esimerkki alasregulaatiosta on solun tietyn reseptorin ilmentymisen väheneminen vasteena molekyylin, kuten hormonin tai hermovälittäjäaineen aktivoitumiselle, mikä vähentää solun herkkyyttä ko. molekyylille. Tämä on esimerkki paikallisesti toimivasta ( negatiivisen palautteen) mekanismista.
Esimerkki ylisääntelystä: sellaisille ksenobiottisille molekyyleille kuin dioksiinille altistettujen maksasolujen vasteena solut lisäävät sytokromi P450 -entsyymien tuotantoa , mikä puolestaan lisää näiden molekyylien hajoamista.
Kaikilla elävillä soluilla on kyky vastaanottaa ja käsitellä solukalvojen ulkopuolelta tulevia signaaleja. Tämän he tekevät reseptoreiksi kutsuttujen proteiinien avulla. Reseptorit sijaitsevat solun pinnalla plasmamembraaniin upotettuna. Kun solunulkoiset signaalit ovat vuorovaikutuksessa reseptorin kanssa, ne ohjaavat solun tekemään jotain, kuten jakautumaan, kuolemaan, tuottamaan proteiineja tai pääsemään energiaravinteita soluun jne. Esimerkiksi insuliinimolekyylin kiinnittyminen insuliinireseptoriin päästää glukoosimolekyylin soluun.
Solun kyky reagoida kemialliseen viestiin riippuu kyseiselle viestille viritettyjen reseptorien läsnäolosta. Mitä enemmän reseptoreita solulla on viritetty ko. signaaliin, sitä vahvemmin solu reagoi siihen. Esimerkiksi insuliiniresistenssissä insuliinireseptorit eivät ole virittyneet, joten solu reagoi heikosti insuliiniin, mikä puolestaan vaikuttaa solun glukoosinottoon ja energian saantiin.
Reseptorit luodaan tai ekspressoidaan solun DNA:n ohjeista, ja niitä voidaan lisätä tai säätää ylöspäin (ylösreguloida), kun signaali on heikko, tai alasreguloida, jos signaali on voimakas.
Niiden tasoa voidaan säätää myös ylös tai alas moduloimalla järjestelmiä, jotka hajottavat reseptoreita, kun solu ei enää tarvitse niitä. Reseptoreiden alasregulointia voi tapahtua myös silloin, kun reseptorit on altistettu kroonisesti liialliselle määrälle ligandia joko endogeenisistä välittäjistä tai eksogeenisista lääkkeistä. Tämä johtaa ligandin aiheuttamaan herkistymiseen tai kyseisen reseptorin sisäistymiseen. Tämä näkyy tyypillisesti eläinhormonireseptoreissa. Reseptorien säätely toisaalta voi johtaa superherkistettyihin soluihin, varsinkin kun toistuva altistuminen antagonistiselle lääkkeelle tai pitkäaikainen ligandin puuttuminen.
2.4. TOR-signaalireitti ja ravitsemus
Elinten ja koko kehon tasolla TORC1 / S6 K1 -signaalireitti säätelee glukoosin homeostaasia, insuliiniherkkyyttä, rasvasolujen aineenvaihduntaa, kehon massa- ja energiatasapainoa, kudosten ja elinten kokoa, oppimista, muistinmuodostusta ja ikääntymistä [57].
TOR (rapamysiinin* kohde) on proteiinikinaasi, joka toimii solujen kasvun ja ikääntymisen keskusohjaimena [43, 44]. TOR-signalointireitin inaktivointi edistää autofagiaa ja pidentää elinikää [45].
*Elimistön vanhenemista on koe-eläimillä pystytty hidastamaan rapamysiinilla. Rapamysiini vähentää elimistön solujen energiankulutusta. Tämä vaikuttaa samalla tavalla kuin ravinnon energiamäärän rajoittaminen. Jos energiaa on puutteellisesti tarjolla, elimistön solujen aineenvaihdunta hidastuu ja samalla niiden elinikä pitenee. Toisaalta rapamysiini heikentää immuunivastetta ja altistaa infektioille.
TOR havaittiin ensin hiivassa, mutta se tunnistettiin myös muissa eukaryooteissa, kuten nisäkkäillä ( TOR tai mTOR). In vivo mTOR esiintyy kahdessa multiproteiinikompleksissa, mTORC1 ja mTORC2.
mTORC1 toimii ravinteiden energia-redoksianturina* ja moduloi proteiinisynteesiä. Siksi alkupään tekijät, jotka stimuloivat tämän kompleksin aktiivisuutta, ovat insuliini ja muut kasvutekijät, aminohapot (esim. leusiini) ja stressi (lämpötilan muutos, kofeiini, oksidatiivinen stressi).
* redox; reduction-oxidation, redoksi; hapetus-pelkistys-reaktio
Kofeiini, hypoksia (happivaje) ja DNA-vauriot estävät mTORC1-aktiivisuutta. TORC1-aktiivisuuden ylävirran säätimet ovat AGC-kinaasiperhe (esim. PKA; PKG ja PKC), jotka aktivoituvat fosforylaatiolla [46]. Nisäkkäillä mTORC1-kohteet ovat S6 K1 ja eukaryoottinen aloituskerroin (4E-BP1) [47–52].
S6 K1:n mTORC1-välitteinen fosforylaatio edistää proteiinisynteesiä ja 4E-BP1-fosforylaatio edistää ribosomien lokalisoitumista mRNA:iden korkkirakenteeseen. MTORC1: n fosforyloivaa aktiivisuutta säätelee sen liittyminen RAPTOR-proteiiniin (mTOR:n säätelyyn liittyvä proteiini) [53, 54].
Korkeat energia- tai ATP-tasot aktivoivat mTORC1:n fosforyloimalla ja siten estäen TSC1-TSC2-kompleksin, kuten Loewith ja Hall ovat osoittaneet [43]. Tämä kompleksi on GTPaasia aktivoiva proteiini, joka modifioi toisen GTPaasi RHEB: n GTP:hen sitoutuneeksi tilaksi. RHEB sitoutuu ja aktivoi GTP:hen sitoutuneen tilan suoraan mTORC1: n ja antaa mTORC1: n fosforyloitua alavirran kohteisiin [55].
Alhainen soluenergia (korkeat AMP-tasot) tai alhaiset ravinnetasot aktivoivat yhdessä tuumorisuppressorikinaasin LBK1 kanssa AMPK:n. Aktivoitu AMPK fosforyloi sekä TSC2:n että RAPTORin ja estää siten mTORC1-aktiivisuuden kahdella reitillä [56].
Hiivassa TORC1 edistää proteiinisynteesiä, ribosomien biogeneesiä, säätelee solusyklin ja solukoon välistä suhdetta, edistää solukasvua estämällä stressivasteita, stimuloi autofagiaa ja säätelee mitokondrioiden toimintahäiriön signaalia ytimeen RTG1-riippuvan negatiivisen säätimen kautta [43, 44].
Elinten ja koko kehon tasolla TORC1 / S6 K1 -signaalireitti säätelee glukoosin homeostaasia, insuliiniherkkyyttä, rasvasolujen aineenvaihduntaa, kehon massa- ja energiatasapainoa, kudosten ja elinten kokoa, oppimista, muistinmuodostusta ja ikääntymistä [57].
Esimerkiksi S6 K1 moduloi mesenkymaalisten kantasolujen erilaistumista adiposyyteiksi. mTORC1 / S6 K1-signalointireitin liiallinen stimulointi liian suurilla määrillä leusiinia äidinmaidonkorvikkeissa voi olla syynä lisääntyneeseen adipogeneesiin ja varhaislapsuuden liikalihavuuteen [58].
mTORC2:n parhaiten ymmärretyt toiminnot ovat aktiinin solurangan solusyklistä riippuvan polarisaation hallinta, endosytoosi ja sfingolipidibiosynteesi [43, 59, 60]. mTORC2: n ylävirran säätimet ovat insuliini ja IGF1 [44, 61].
Ribosomin kypsytystekijä Nip7 vaaditaan mTORC2-kinaasiaktiivisuuteen hiiva- ja nisäkässoluissa [44, 61] ja mTORC2: n substraatit ovat AGC-kinaasiperhe mukaan lukien AKT, SGK1 ja PKC [44, 62]. Esimerkiksi mTORC2 edistää solujen eloonjäämistä AKT:n kautta [63, 64] ja säätelee myös maksan glukoosi- ja lipidimetaboliaa insuliinin indusoiman AKT-signaloinnin kautta [62]. Vaikka TORC1:n ja TORC2:n signalointireitit ovat jossain määrin erillisiä, niillä on yhteistyöfunktio koordinoida kasvua, mitoosia ja solukoon hallintaa.
Esimerkiksi TORC2 aktivoi TORC1:n AKT-signalointireitin kautta. TORC1-aktivaatio stimuloi anabolisia solureittejä ja TORC1-esto stimuloi katabolisia solupolkuja [65]. TORC1- ja TORC2-signalointireittien herkkyys voi yleensä olla paitsi solukudosspesifinen myös TORC-isoformista riippuvainen. Esimerkiksi mTORC2: n aktiivisuus riippuu nisäkkään stressiaktivoituneen proteiinikinaasia vuorovaikutuksessa olevan proteiinin (mSin1) isoformista, joka muodostaa tämän multiproteiinikompleksin [66].

3. Ravinto ja vanheneminen
Oletus, että nisäkkäiden elinikää voitaisiin pidentää merkittävästi ruokavalion muutoksilla, vahvistettiin jyrsijätutkimuksessa, jonka toteuttivat McCay ym. vuonna 1935 [67].
Rotat kasvavat koko ikänsä. Yksi tämän tutkimuksen tavoitteista oli määrittää kasvun hidastumisen vaikutus molempia sukupuolia olevien rottien eliniän pituuteen. Kasvu hidastui rajoittamalla ravinnosta saatavan energian määrää tasolle, joka on tarpeen rottien pitämiseksi vakailla ruumiinpainotasoilla vieroituksen aikana tai 2 viikkoa vieroituksen jälkeen.
Kokeessa huolehdittiin kaikkien muiden ruokavalion ainesosien riittävästä saannista. Ruokavalion energiamäärän rajoittaminen pidensi rottien elinikää. Ruokavalion rajoittamisen vaikutus elinaikaan oli kuitenkin selvempi uros. kuin naarasrotilla [67].
Yhteenvetona voidaan todeta, että tämä peruskokeilu viittaa siihen, että elinikää voidaan pidentää ruokavalion sisältämän energiamäärän rajoittamisella ilman aliravitsemusta. Aliravitsemuksella voi olla päinvastainen vaikutus [1].
Suositeltava ravintoprotokolla on energiansaannin rajoittaminen siten, että kalorirajoitus ei aiheuta välttämättömien ravintoaineiden puutostiloja tai aliravitsemusta. CR tarkoittaa kalorien saannin rajoittamista 10–30% verrattuna energian normaaliin saantiin. Energiansaannin hallitun rajoittamisen on osoitettu parantavan kaiken ikäisten terveyttä ja hidastavan myös ikääntymistä monilla tutkituilla eukaryooteilla [68].
Energian rajoittamisen elinikää pidentävien vaikutusten merkitystä kädellisille on tutkittu mm. reesusapinoilla. Eläinten lähtötason kaloreiden saantia laskettiin asteittain 10% kuukaudessa lopulliseen 30% energian rajoitukseen, joka säilyi kokeen ajan. CR:n vaikutus verrokkeihin arvioitiin vertaamalla kuolleisuuden viivästymistä ja joidenkin ihmisillä yleisimmin esiintyvien ikään liittyvien sairauksien (esim. diabetes, syöpä, sydän- ja verisuonitaudit ja aivojen atrofia) puhkeamista.
Tutkimuksen johtopäätökset olivat, että kaloreiden rajoittaminen alensi ikääntymiseen liittyvien kuolemien esiintyvyyttä (50% kontrolliruokituilla eläimillä verrattuna 20% CR-ruokituilla eläimillä) ja alensi myös diabeteksen, syövän, sydän- ja verisuonitautien sekä aivojen atrofian ilmaantuvuutta [68 ].
Tältä pohjalta voidaan kysyä: liittyykö okinawalaisten pitkäikäisyys niukkaan energiansaantiin?

4. Kaloreiden rajoittamisen vaikutukset ihmisillä
Perusoletus, jonka mukaan kalorirajoitus voi pidentää keskimääräistä ja enimmäisikää ja viivästyttää ikään liittyvien muutosten alkamista, on osoitettu monissa organismeissa hiivasta, matoihin ja kärpäsistä nisäkkäisiin [69–71].
Kehittyneemmillä nisäkkäillä kalorirajoitus viivästyttää monia ikääntymiseen liittyviä sairauksia, kuten syöpää, diabetesta, ateroskleroosia, sydän- ja verisuonitauteja sekä neurodegeneratiivisia sairauksia [68, 72–74]. Näiden sairauksien ilmaantuvuus kasvaa iän myötä ja ne vaikuttavat merkittävästi kuolleisuuteen. Energiansaannin rajoittaminen voi pidentää elinikää lisäämällä kehon yleistä terveydentilaa ja tarjoamalla epäspesifistä vastustuskykyä kroonisille sairauksille ja aineenvaihdunnan häiriöille [68, 72–74].
Lopullista kysymystä, miten kaloreiden rajoittaminen vaikuttaa ihmiskehoon, tutkittiin kuitenkin rajoitetulla määrällä kokeita [73–93]. Tutkimuksella CR-vaikutuksista ihmisen pitkäikäisyyteen liittyy eettisiä ja logistisia haasteita, koska kehittyneiden maiden väestön keskimääräinen elinikä on lähes 80 vuotta. Siksi ihmisen tutkimuksissa keskitytään mittaamaan kaloreiden rajoittamiseen liittyviä muutoksia, jotka voivat hidastaa ikääntymistä ja kroonisten sairauksien etenemistä, mikä pidentää elinikää.
Vakuuttavin näyttö siitä, että CR:llä voi olla positiivinen vaikutus ihmisiin, saatiin Fontanan ym. kokeilla ja kattavalla arvioinnilla kalorien saannin rajoittamisen pitkäaikaisista vaikutuksista (CALERIE Phase 1, josta puhutaa ensimmäisellä videoluennolla) ja saaduista tiedoista. Caloric Restriction Society (kuten jäljempänä keskustellaan). Fontana ym. arvioivat 6 vuoden pituisen CR-ruokavalion vaikutusta ateroskleroosin riskitekijöihin mies- ja naispuolisilla aikuisilla (ikä 35–82-vuotiaat) ja heitä verrattiin iältään vastaaviin terveisiin tyypillistä amerikkalaista ruokavaliota noudattaviin ihmisiin (kontrolliryhmä).
Seerumin kokonaiskolesterolitaso ja LDL-kolesterolitasot, kokonaiskolesterolin suhde suurtiheyksiseen lipoproteiinikolesteroliin (HDL), triglyseridit, paastoglukoosi, paastoinsuliini, C-reaktiivinen proteiini (CRP), verihiutaleista johdettu kasvutekijä AB sekä systolinen ja diastolinen verenpaine olivat kaikki selvästi pienempiä kaloreita rajoittavassa ryhmässä kuin kontrolliryhmässä.
HDL-kolesteroli oli korkeampi kaloreiden rajoittamisen jälkeen. CR-ryhmän henkilöiden lääketieteelliset tiedot osoittivat, että ennen kaloreiden rajoittamisen aloittamista heillä oli seerumin lipidi-lipoproteiini- ja verenpainetasot samalla tasolla kontrolliryhmän tyypillistä amerikkalaista ruokavaliota noudattavien henkilöiden kanssa ja samanlainen kuin vertailuryhmässä. Tutkimuksen johtopäätös oli, että pitkäaikainen kaloreiden rajoittaminen voi vähentää ateroskleroosin riskitekijöitä [74].
(a) Pitkäaikaisen 20%:n kalorirajoituksenn ja (b) 20%:n lisääntyneen energiankulutuksen (IEE) aiheuttaman rasvanpudotuksen vaikutusta sepelvaltimotaudin (CHD) riskitekijöihin arvioitiin yhden vuoden satunnaistetussa kontrolloidussa tutkimuksessa 48 ei-lihavalla mies- ja naishenkilöllä.
Kaloreiden rajoittamisen (a) ja liikunnan (b) vaikutus rasvakudoksen vähentymiseen olivat määrällisesti vastaavia. Kaloreiden rajoittaminen ja liikunta vaikuttivat yhtäläisesti myös CHD-riskitekijöiden, kuten plasman LDL-kolesterolin, kokonaiskolesteroli / HDL-suhteen ja CRP-pitoisuuden laskuun.
Tutkimuksen tekijät päättelivät, että saman suuruusluokan pitkäaikainen kaloreiden rajoitus tai liikunnan avulla saavutettava lisääntynyt energiankulutus (IEE) johtavat merkittävään ja yhtäläiseen sydänterveyden riskitekijöidenterveysmarkkereiden kohenemiseen normaalipainoisilla ja ylipainoisilla keski-ikäisillä aikuisilla [83].
Vuoden mittainen 20% kaloreita rajoittavan ruokavalion ja 20% liikunnan avulla energiankulutusta lisäävän IEE:n vaikutukset arvioitiin DNA:n ja RNA:n oksidatiivisten vaurioiden osalta valkosolu- ja virtsa-analyyseillä normaali- ja ylipainoisilla aikuisilla. Molemmat interventiot vähensivät merkittävästi sekä DNA:n että RNA:n oksidatiivisia vaurioita valkosoluissa verrattuna lähtötasoon.
Virtsasta tutkittujen DNA:n ja RNA:n oksidatiiviset vauriot eivät kuitenkaan eronneet lähtötasosta kummankaan interventio-ohjelman jälkeen. Tutkimuksen johtopäätös oli, että sekä kaloreiden rajoittaminen että IEE vähentävät systeemistä oksidatiivista stressiä, mikä heijastuu vähentyneinä DNA:n ja RNA:n hapettumisvaurioina [85].
CALERIE on kansallisen ikääntymislaitoksen käynnistämä tutkimusohjelma, johon osallistuu kolme tutkimuskeskusta. CALERIE-vaiheeseen sisältyi kolme pilottitutkimusta sen selvittämiseksi, voidaanko pitkäaikaisen (6–12 kuukautta) 20–25%:n kaloreiden rajoittamisen vaikutuksia tutkia normaalisti elävien ei-lihavien aikuisten osalta ja arvioida kaloreiden rajoittamisen adaptiivisia vasteita.
Tämän satunnaistetun kontrolloidun kliinisen tutkimuksen johtopäätökset olivat, että kaloreita rajoittavilla koehenkilöillä oli alempi ruumiinpaino, vähentynyt kehon ja sisäelinten rasvapitoisuus, pienempi aktiivisuusenergiankulutus, parantuneet paastoinsuliinipitoisuudet, parantuneet sydän- ja verisuonitautien riskiä ennustavat markkerit (LDL, HDL-suhde ja CRP), eikä muutoksia luun tiheydessä verrokkeihin verrattuna [76, 77, 83, 86, 92].
Käynnissä olevan CALERIE-tutkimuksen toisen vaiheen tavoitteena on testata, johtaako 2 vuoden vapaa 25% kaloreiden rajoittaminen samanlaisiin suotuisiin vaikutuksiin, kuin eläinkokeissa havaitut vaikutukset [91].
Caloric Restriction Societyn (CRS) jäsenet rajoittavat energian saantia olettaen, että tämä viivästyttää sekundaarisestä ikääntymisestä johtuvia sairausprosesseja ja hidastaa primaarista ikääntymistä.
Verrattuna saman ikäisiin tyypillistä amerikkalaista ruokavaliota noudattaviin amerikkalaisiin, CRS-jäsenillä (keski-ikä 50 ± 10 vuotta) oli alhaisempi painoindeksi, pienempi kehon rasvaprosentti, merkittävästi alemmat arvot seerumin kokonaiskolesterolille, LDL-kolesterolille, kokonaiskolesterolille / LDL:lle ja korkeampi HDL-kolesteroli. Myös plasman paastoinsuliinin ja glukoosin pitoisuudet olivat merkittävästi alhaisemmat kuin ikäryhmän verrokkiryhmässä.
Vasemman kammion diastolinen toiminta CRS-jäsenillä oli samanlainen kuin noin 16 vuotta nuoremmilla henkilöillä. Kaloreita rajoittava ruokavalio hiljensi kroonista tulehdusta ja tämä ilmeni plasman CRP:n ja tuumorinekroositekijän-alfan (TNFα) merkittävästi alhaisemmissa tasoissa [74, 78, 84].
Ikääntyminen liittyy sykevälivaihtelun (HRV) asteittaiseen heikkenemiseen. Tämä osoittaa sydämen autonomisen toiminnan heikkenemistä ja yleisesti heikentynyttä terveyttä.
Energian saannin rajoittaminen 30 %:lla vaikuttaa myönteisesti sydämen autonomiseen toimintaan. Kaloreita rajoittavassa ryhmässä oli alempi syke ja huomattavasti korkeammat HRV-arvot. Tutkijat arvelevat, että kaloreiden rajoittaminen palauttaa tasapainon sydämen taajuuden sympaattisen / parasympaattisen moduloinnin välillä parasympaattisen ajon eduksi, mikä lisää sykkeen vuorokausivaihtelua [93].

5. Kaloreiden rajoittamisen vaikutukset solutasolla
Suurin osa ikään liittyvistä muutoksista geeniekspressiossa on melko vähäisiä ja kudosspesifisiä [94]. Silti kudosspesifisistä eroista iän vaikutuksessa geenitranskriptioon ikääntymisnopeus kudoksissa vaikuttaa olevan koordinoitua, mikä viittaa systeemisten tekijöiden merkitykseen ikääntymisprosessin koordinoinnissa koko kehon tasolla [95].
Yleisimpiä ikään liittyviä muutoksia ovat lisääntynyt tulehdukseen ja immuunivasteisiin liittyvien geenien ilmentyminen ja mitokondrioiden (MTH) energia-aineenvaihdunnan heikkeneminen. Kaloreiden rajoittamiseen liittyvien geenien vähentynyt ilmentyminen estää suurimman osan näistä ikään liittyvistä muutoksista geeniekspressiossa [96, 97 ]. Kaloreiden rajoittamisen arvellaan vastaavan ikään liittyviä muutoksia moduloimalla mTOR-signalointireitti, IGF1 / insuliinisignalointi, adiponektiiniekspressio, DNA-metylaatio ja histoniasetylointi ja deasetylointi.
5.1. Kalorirajoituksen vaikutus adiponektiinien eritykseen
Johdonmukainen muutos energiaa rajoittavan ruokavalion aikana on kehon rasvan väheneminen (ts. valkoisen rasvakudoksen väheneminen). Valkoinen rasvakudos ei ole vain lipidien varastointipaikka, vaan sillä on tärkeä rooli verensokerin homeostaasissa, immuuni- ja tulehdusreaktioissa, jotka välittyvät adiposyytteistä peräisin olevista solusta soluun signaloivista molekyyleistä, adipokiineista (esim. adiponektiini) [98 , 99].
Siksi rasvakudos voi olla tärkeä tekijä ikääntymiseen ja kaloreiden rajoittamiseen (CR) liittyvissä aineenvaihdunnan muutoksissa. Adiponektiinin eritystä lisää vähentynyt kalorien saanti.
Adiponektiini vähentää sekä insuliinia että IGF1:tä, jotka vastaavasti vähentävät adiponektiinin synteesiä. Poikkileikkaustutkimukset osoittavat tasaisen käänteisen korrelaation plasman insuliini- ja adiponektiinipitoisuuksien välillä. Adiposyyttien (rasvasolujen) koon kasvu vähentää myös adiponektiinin eritystä [100]. Adiponektiini edistää rasvahappojen hapettumista rasvakudoksessa ja vähentää lipidien kertymistä muihin ääreiskudoksiin [101]. Kaloreiden rajoittaminen lisää veren adiponektiinipitoisuutta [102].
Ihmisillä tämä hormoni tukahduttaa aineenvaihdunnan häiriöt, jotka voivat johtaa tyypin 2 diabetekseen, lihavuuteen, ateroskleroosiin tai metaboliseen oireyhtymään [103–105]. Adiponektiini säätelee mitokondrioiden energiantuotantoa AMPK:n kautta.
AMPK:lla on monia toimintoja. Se säätelee ylöspäin (ylösreguloi) glukoosin imeytymistä soluun, rasvahappojen β-oksidaatiota, glukoosin kuljettaja 4:n (GLUT4) ilmentymistä ja mitokondrioiden energiantuotantoa.
AMPK-entsyymillä on ”energiaa tunnistava kyky”. Se se reagoi solunsisäisen AMP / ATP-suhteen vaihteluihin. Esimerkiksi ihmisen ”myo-putkien”* (myotube, en löytänyt suomennosta tälle sanalle) adiponektiinihoito johtaa AMPK:sta riippuvaan MTH-biogeneesin lisääntymiseen ja vähentää reaktiivisten happilajien (ROS) tuotantoa [106].
”Myotubes have rows of centrally located nuclei and peripheral masses of forming contractile myofilaments that soon become oriented into sarcomeres and myofibrils with restoration of cross-striations in the immature myofibers.”
AMPK säätelee MTH-energiantuotantoa aktivoimalla peroksisomiproliferaattorilla aktivoidun reseptori-gamma-koaktivaattori 1-alfan (PGC1-α) suoraan tai endoteelin typpioksidisyntaasin (eNOS) ja NAD-riippuvaisen deasetylaasi-sirtuiini1:n (eli SIRT1:n) kautta säännön 2 homologi 1) signalointireitillä.
AMPK: n lisääntyneellä aktiivisuudella kaloreita rajoittavan ruokavalion aikana on myös sydäntä suojaava kardioprotektiivinen vaikutus [102]. Lisääntynyt AMPK-aktiivisuus stimuloi myös eNOS-aktiivisuutta ja vähentää siten aivojen iskeemisen vaurion todennäköisyyttä [107]. Muita kardioprotektiivisia vaikutuksia, joita välittää lisääntynyt adiponektiinin eritys kaloreita rajoittavan ruokavalion aikana, ovat (a) TNF-α:n erittymisen estäminen ja (b) adheesiomolekyylien synteesin esto endoteelisoluissa. Jälkimmäinen estää monosyyttien kiinnittymisen endoteelisoluihin ja viivästyttää ateroskleroosin etenemistä.
Adiponektiinimoduloidut tulehdusvasteet johtuvat TNF-α :n (tuumorinekroositekijä-alfa on systokiini, joka liittyy systeemiseen tulehdukseen) erityksen estämisestä monosyytistä / makrofagista ja vaahtosolusta [108–110]; tämä voi selittää tulehdusproteiinin CRP:n pienentyneen plasmakonsentraation ihmisillä, jotka noudattavat niukasti energiaa sisältävää ruokavaliota.
5.2. Energian rajoittamisen vaikutus insuliini/IGF1-signalointiin
Insuliiniresistenssi on tunnettu ikään liittyvä aineenvaihdunnan häiriö, jonka niukkaenerginen ravinto, pätkäpaasto, paasto ja ketogeeninen ruokavalio voivat estää ja parantaa [94].
Kalorirajoituksen on raportoitu vähentävän IGF1:n pitoisuutta hiirillä, mutta ei ihmisillä [111, 112]. Insuliini ja IGF1 estävät FOXO-proteiineja* signalointireitillä, joka sisältää insuliinireseptorisubstraattiproteiineja (IRS), 3-fosfoinositidista riippuvaa proteiinikinaasia-1 (PDPK1) ja fosfatidyylinositoli-3-kinaasia ( PTDINS-3 K), siirtäen siten FOXO:t tumasta.
*FOX (forkhead box) -proteiinit ovat perhe transkriptiotekijöitä, joilla on tärkeä rooli solujen kasvuun, lisääntymiseen, erilaistumiseen ja pitkäikäisyyteen liittyvien geenien ilmentymisen säätelyssä. Monet FOX-proteiinit ovat tärkeitä alkion kehitykselle. FOX-proteiineilla on myös merkittävä transkriptiovaikutus, koska ne kykenevät sitomaan kondensoitunutta kromatiinia solujen erilaistumisprosessien aikana.
FOXO-transkriptiotekijät vaikuttavat ikääntymiseen vasteena ravinnolle ja sen energiapitoisuudelle. Tämän aineenvaihduntareitin puuttuminen nisäkkäillä liittyy lihavuuteen ja insuliiniresistenssiin [113].
Solutyyppispesifisellä tavalla nisäkkään FOXO-tekijät kontrolloivat erilaisia solutoimintoja, mukaan lukien apoptoosi (ohjattu solukuolema), solusykli, erilaistuminen ja DNA-korjaukseen ja oksidatiiviseen stressiresistenssiin liityvien geenien ilmentyminen.
Näiden toimintojen oletetaan olevan perusta FOXO-tekijöiden kyvylle hallita elinkaarta [114]. Mustan teen polyfenolit jäljittelevät insuliinin / IGF1-signalointireitin vaikutuksia FOXO1a-transkriptiotekijään [113] ja FOXO3a-geenin polymorfismit liittyivät ihmisten pitkäikäisyyteen [115].
Kaloreiden rajoittaminen stimuloi FOXO3a:n SIRT1-välitteistä deasetylaatiota, estäen tuman FOXO3a-aktiivisuuden ja estämällä Rho-assosioituneen proteiinikinaasi-1:n ilmentymisen aktivoimalla APP:n ei-amyloidogeenisen α-sekretaasin prosessoinnin ja alentamalla Aβ:n muodostumista. Tämä vähentynyt Aβ-sukupolvi liittyy Alzheimerin taudityyppisen amyloidineuropatologian ja spatiaalisen muistin heikkenemisen estämiseen hiirimallissa [114].
Niukan energiansaasnnin positiivinen vaikutus insuliini / IGF1-signalointireittiin liittyi myös ROS-tuotannon vähenemiseen MTH: ssa [116].
5.3. Energian rajoittamisen vaikutus mTOR-signalointiin
Elinkaaren säätelyä mTOR-signalointireitillä ei täysin ymmärretä. Viimeaikainen kokeellinen työ viittaa kuitenkin siihen, että sillä on keskeinen rooli solun ikääntymisprosessissa [44]. MTOR-signalointireitin estäminen rapamysiinillä pidentää maksimaalista ja mediaaniaikaa hiirillä. Tämä vaikutus havaittiin silloinkin, kun hoito aloitettiin myöhässä, mikä vastaa suunnilleen 60 vuoden ikää ihmisillä [44, 117]. Edellä mainittu, rapamysiinivälitteinen elinajan pidentyminen ei liittynyt muutoksiin sairausmalleissa tai kuolinsyissä, mikä viittaa siihen, että rapamysiini pidentää elinikää hidastamalla ikään liittyvää kudosten ja elinten rappeutumista [44, 117].
mTORC1-esto voi estää kudosten rappeutumisen ja pidentää elinikää parantamalla kantasolujen toimintaa. Esimerkiksi mTORC1-signaloinnin vähentäminen rapamysiinillä palauttaa hematopoieettisten kantasolujen itsensä uudistumisen ja hematopoieettisen toiminnan, parantaa immuniteettia ja pidentää hiirien elinikää [118].
S6 K1 ja 4E-BP1 arvellaan ikääntymisprosessia säätelevän mTORC1-signalointireitin efektoreiksi. Kuten Kapahi et al. on osoittanut, pienentynyt S6 K1 -aktiivisuus pidentää elinikää eri lajeilla, myös hiirillä [119], ja 4E-BP1:n yli-ilmentyminen pidentää elinikää rikkaissa ravinto-olosuhteissa parantamalla mitokondrioiden aktiivisuutta kärpäsillä [120].
mTORC1 voi myös vaikuttaa elinikään sellaisten mekanismien kautta, jotka eivät liity proteiinisynteesin modulointiin; esimerkiksi autofagian stimulaatio mTORC1-eston seurauksena voi edistää pitkäikäisyyttä stimuloimalla soluihin keräätyvien poikkeavien proteiinien ja vaurioituneiden organellien hajottamista ja kierrättämistä. Soluihin kerääntyy ajan myötä erilaisia solun toimintaa heikentäviä kuona-aineita [44].
Esimerkki siitä, kuinka mTORC1-aktiivisuuden säätely vaikuttaa elinikään, nähdään vanhojen hiirten maksan heikentyneenä paaston aiheuttama ketogeneesinä ja lisääntynyneenä mTORC1-aktiivisuutena [121]. Tämä heikentynyt ketogeneesi rajoittaa käytettävissä olevien energiasubstraattien määrää ääreiskudoksiin vähentäen siten organismin mahdollisuuksia selvitä ravinnon puutteen aikana. Kaloreiden rajoittaminen vähentä ikään liittyvää MTH-toiminnan heikkenemistä [69].
Kaloreiden rajoittamisen vaikutukset MTH:een voidaan välittää myös mTOR-signalointireitillä, koska mTOR on välttämätön mitokondrioiden oksidatiivisen toiminnan ylläpitämiselle [122]. Kahta S6 K1- ja 4E-BP1-riippumatonta mTOR / MTH-signalointireittiä on ehdotettu: TORC1-YY1-PGC-1α-kompleksia, [122] joka on osoitettu hiirimallissa ja TORC1-säännelty BCL-XL:n ja VDAC1:n kompleksi, joka sijaitsee mitokondrioiden ulkokalvolla [123].

5.4. DNA-metylaatio kaloreita rajoittavalla ruokavaliolla
Ikääntymisprosessiin liittyy vähitellen heikkenevä solujen homeostaasi ja geeniekspressioon muutokset [124]. Vanheneminen aiheuttaa merkittävän muutoksen 5-metyylisytosiinin (DNA-metylaation tuote) jakautumisessa genomiin ja yleisemmin vähenevä genomin DNA-metylaati [124–130].
Joidenkin spesifisten geenien promoottorialueilla on taipumus siirtyä metyloitumattomasta metyloituneeseen tilaan, mikä johtaa geenien hiljentämiseen (esim. tuumoripromoottorit tai ikääntymiseen liittyvät geenit, kuten RUNX3 ja TIG1 [129, 131]). Yhteenvetona voidaan todeta, että ikääntymisprosessi liittyy yleisesti vähentyneeseen, mutta paikallisesti lisääntyneeseen DNA-metylaatioon [132].
Kaloreiden rajoittamisen oletetaan viivästyttävän ikääntymisprosessia kääntämällä ikääntymiseen liittyvät DNA:n metylaatiomuutokset lisäämällä siten genomista vakautta [133, 134]. Kaloreiden rajoittaminen esimerkiksi nosti proto-onkogeenin RAS:n metylaatiotasoa rotamallissa verrattuna ad libitum -syötettyihin eläimiin [135].
Hypermetyloidun geenipromoottorin tunnistaa usein transkriptionaalisista repressorikomplekseista, mikä johtaa näiden onkogeenien ilmentymisen vaimentamiseen ja mikä edelleen osaltaan selittää kalorirajoituksen syöpiä ehkäisevää vaikutusta [132].
Niukkaenergisen ravinnon in vitro -solumallissa geenin promoottorissa (tuumorisuppressori ja ikääntymiseen liittyvä geeni) E2F-1:n sitoutumiskohta hypermetyloitiin. Tämä DNA:n hypermetylaatio esti E2F-1:n (aktiivisen transkriptiotekijän) pääsyn promoottoriin, mikä johti alasregulaatioon ja mikä osaltaan myötävaikuttaa kalorirajoituksen indusoimaan elinkaaren pidentymiseen [136].
Lihavuus on yleinen metabolinen häiriö. Se liittyy läheisesti kiihtyneen ikääntymisen ja lisää kuolleisuutta diabetekseen, hypertensioon, syöpiin ja sydän – ja verisuonitauteihin [137]. Siksi kaloreiden rajoittamisen ikääntymistä hidastavilla anti-aging vaikutuksilla on vaikutusta lihavuuden etenemiseen. Energiansaannin rajoittamista käytetään kliinisissä painonhallintatoimissa [138].
Lihavien ihmisten noudattaman kaloreiden rajoittamisen tutkimus osoitti, että vähäkaloriset ruokavaliot aiheuttavat DNA-metylaation muutoksia spesifisissä lokuksissa ATP10A, WT1 ja TNF-α, Näitä muutoksia voidaan käyttää kaloreiden rajoittamisen vasteen varhaisina indikaattoreina [139–141]. Lisätutkimukset ihmisillä ovat välttämättömiä niiden DNA-metylaatio-ohjattujen ehdokasgeenien joukon luonnehtimiseksi, jotka voivat olla läheisessä korrelaatiossa metabolisten reittien kanssa [132].
5.5. Histonien translaation jälkeinen muuntaminen kaloreita rajoittavalla dieetillä
5.5.1. Histoniasetylointi / deasetylointi
Histonimodifikaatiot liittyvät geeniaktivaatioon tai geenirepressioon. Histonipään sisällä olevien modifikaatioiden yhdistelmä muuttaa nukleosomien konfiguraatiota vaihtamalla kromatiinin joko tiivistetyksi (tiiviisti kiinni) tai rennoksi kokoonpanoksi (löysästi auki) [142].
Siksi histonimodifikaatiot määrittävät kromatiinin (tiukasti kiinni: löysästi auki) -suhteen ja siten geeniaktiivisuuden asteen tietyllä DNA-alueella. Esimerkiksi deasetyloidulla histonilysiinitähteellä on positiivinen varaus, joka houkuttelee negatiivisesti varautuneita DNA-säikeitä ja tuottaa kompaktin kromatiinitilan, joka liittyy transkriptiorepressioon. Vaihtoehtoisesti histoniasetylaatio poistaa positiivisen varauksen ja johtaa avoimeen kromatiinirakenteeseen, joka edistää geenitranskriptiota [132].
HDAC-aktiivisuus lisääntyy niukkaenergisen ruokavalion aikana, joten elimistön yleinen deasetylaatio voi olla sellainen suojamekanismi ravitsemusstressiä vastaan, joka voi vaikuttaa ikääntymisprosesseihin [136].
Kaloreita rajoittamalla esimerkiksi HDAC1:n lisääntynyt aktiivisuus ihmisen telomeraasikäänteiskopioijaentsyymin (hTERT) geenien promoottorialueilla, joista ensimmäinen on tuumorisuppressori monissa syövissä ja jälkimmäinen keskeinen ikääntymiseen vaikuttava telomeraasiaktiivisuuden säätelijä, johtaa näiden kahden geenin ilmentymiseen ja hyödyllisiin muutoksiin, jotka myötävaikuttavat pitkäikäisyyteen [136, 143, 144].
Useita HDAC-perheitä on tunnistettu. Näihin kuuluu mm. luokan III NAD+ -riippuvat HDAC:t, kuten Sirtuin1. Sirtuin1 (SIRT1 nisäkkäillä) ja sen ortologit muilla lajeilla (esim. Sirtuin2 hiivassa) ovat tärkeitä ikääntymisen säätelijöitä niukkaan energiansaantiin liittyvän eliniän pidentämisessä [145–149].
SIRT1:n entsymaattinen aktiivisuus riippuu NAD+ / NADH -suhteesta, joka on keskeinen indikaattori hapenkulutukselle. Tämä viittaa siten siihen, että tämä proteiini reagoi solujen metaboliseen tilaan. SIRT1:n roolia kaloreiden rajoittamisen ja eliniän pidentämisen yhtenä säätelijänä ja selittäjänä tukevat useat eläinmallit, ihmiskohteet ja in vitro solujärjestelmät [136, 145, 146, 148–154].
Kaloreiden rajoittaminen indusoi SIRT1-ekspression useissa hiirien tai rottien kudoksissa [146]. SIRT1:n oletetaan välittävän niukan energiansaannin aiheuttamia aineenvaihdunnan muutoksia ja ikääntymisen hidastumiseen liittyviä prosesseja:
(a) lisäämällä stressiresistenssiä p53:n ja FOXO:n negatiivisella säätelyllä [155–159]
(b) aloittamalla sarjan endokriinisiä vasteita, kuten adipogeneesin* vähentämisen ja insuliinin erityksen estämisen haiman β-soluista säätelemällä tärkeitä aineenvaihduntaan liittyviä geenejä, kuten peroksisomiproliferaattorilla aktivoituja G-reseptorin koaktivaattoria lα (PGC-1α) [160, 161].
* Adipogeneesi valmistaa adiposyyttejä (rasvasoluja) kantasoluista.
Vaikka SIRT1 on luokiteltu HDAC:ksi, se deasetyloi myös nonhistonisubstraatit [146, 152], kuten keskeiset transkriptiotekijät (esim. FOXO), säätelyproteiinit (esim. P53,) ja DNA:n korjausproteiinit (esim. Ku70), jotka vaikuttavat ikääntymisen hidastumiseen niukasti energiaa sisältävällä ruokavaliolla.
Esimerkiksi p53:n vähentäminen SIRT1-deasetylaatiolla voi vaikuttaa elinikään estämällä solujen apoptoosia (ohjattua kuolemaa) ja replikatiivisia vanhenemisprosesseja [155–157, 162–164]. FOXO-proteiini voidaan deasetyloida suoraan SIRT1:llä lysiinitähteissä ja sen ilmentyminen vähenee, mikä tukahduttaa FOXO-välitteisen apoptoosin [158,159].
DNA:n korjausproteiini, Ku70*, voi deasetyloida SIRT1:n, antaen sen inaktivoida proapoptoottinen tekijä BAX ja estäen siten apoptoosia [165, 166].
* Ku on dimeerinen proteiinikompleksi, joka sitoutuu DNA:n kaksoisjuosteiden päihin ja jota tarvitaan DNA:n ei-homologisen päätyliittymisreitin (NHEJ) korjaamisen. Ku on evolutiivisesti säilynyt bakteereista ihmisiin. Eukaryoottinen Ku on kahden polypeptidin, Ku70 (XRCC6) ja Ku80 (XRCC5), heterodimeeri. Kaksi Ku-alayksikköä muodostavat korin muotoisen rakenteen, joka kietoutuu DNA-kaksoisjuosteen-päähän. Monimutkaisemmissa eukaryooteissa Ku muodostaa kompleksin DNA-riippuvaisen proteiinikinaasikatalyyttisen alayksikön (DNA-PKcs) kanssa muodostaen täydellisen DNA-riippuvaisen proteiinikinaasin, DNA-PK: n. Ku:n uskotaan toimivan molekyylitelineenä, johon muut NHEJ:ssä mukana olevat proteiinit voivat sitoutua. Ku70- ja Ku80-proteiinit koostuvat kolmesta rakenteellisesta domeenista. N-terminaalinen domeeni on alfa / beeta-domeeni. Ku70:n ja Ku80:n keskeinen domeeni on DNA:ta sitova beeta-tynnyrialue.
Ku70 on sykliiniriippuvainen kinaasin estäjä, tärkeä kasvainsuppressoijaproteiini ja potentiaalisesti ikääntymisen biomarkkeri, koska sitä kertyy merkittävästi ikääntymisprosessien aikana [167–171]. Kalorirajoituksen aktivoima SIRT1 voi sitoutua suoraan promoottoriin ja vähentää sen ilmentymistä deasetylaatioefektin kautta, mikä osaltaan viivästyttää ikääntymisprosessia ja pidentää elinikää ihmissoluissa in vitro [153].
Kuten aiemmin todettiin, SIRT1 säätelee myös metaboliareiteissä mukana olevien geenien ilmentymistä. PGC-1α on keskeinen glukoneogeneesin ja rasvahappojen hapettumisen säätelijä [160, 161], ja sitä säätelevät niukan energiansaannin aikana SIRT1-välitteinen deasetylaatio, mikä lisää sen kykyä koaktivoida HNF4a:ta (transkriptiotekijä, joka edistää glukoneogeenisten geenien ilmentymistä ja tukahduttaa geenit mukana glykolyysissä) [147, 152]. Yhteenvetona voidaan todeta, että SIRT1:llä on keskeinen rooli epigeneettisten ja geneettisten reittien keskinäisessä viestinnässä [132].
5.5.2. Histonien metylaatio
Toisin kuin histoniasetylaatio, joka liittyy avoimeen kromatiinitilaan ja sen jälkeiseen geeniaktivaatioon, eri tavoin metyloiduilla histonimuodoilla on spesifit assosiaatiomallit spesifisten proteiinien kanssa. Ne tunnistavat nämä markkerit ja johtavat siten geenien hiljentämiseen tai aktivaatioon [132].
Histonilysiinitähteet voivat olla mono-, di- tai tri-metyloituja, mikä johtaa joko geenin aktivaatioon tai repressioon riippuen modifioidusta lysiinitähteestä [172, 173].
Niukka energiansaanti indusoi histonimetylaatiomodifikaatiot, kuten di- tai tri-metyloitu histoni H3 lysiinitähteissä 3 ja 4, säädellen keskeisten ikääntymiseen liittyvien geenien ja hTERT:n ilmentymistä ja myötävaikuttivat siten kalorirajoituksen aiheuttamaan ihmissolujen elinikän pidentymiseen [136, 153].
5.6. Kaloreiden rajoittamisen vaikutus miRNA-ekspressioon
miRNA-ilmentymismallit muuttuvat iän myötä. Jotkut miRNA:t ovat alasreguloituja ja toiset ylössäänteltyjä. Ihmisen veren perifeeristen mononukleaaristen solujen 800 miRNA:n ilmentymisprofiilianalyysi osoitti, että suurin osa miRNA:ista väheni määrällisesti, mukaan lukien syövän kehitykseen osallistuvat miRNA:t [174].
Koska ihmisen kasvaimiin liittyy usein miRNA:iden yleinen alasregulointi, raportoitu ikään liittyvä yleinen miRNA:n väheneminen voi lisätä solumuunnoksen ja kasvaimen syntymisen riskiä ja siten vähentää elinikää. Näiden jälkimmäisten miRNA:iden väheneminen iäkkäillä liittyi myös kohdeproteiinien fosfatidyylinositoli-3-kinaasin, kantasolutekijäreseptorin (c-KIT) ja histoni H2A:n lisääntyneeseen ilmentymiseen [174].
Eläintutkimukset tukevat myös miRNA:iden roolia ikääntymisessä. Esimerkiksi kontrolleihin verrattuna C. elegans -mutaatioissa, joissa on poistettu miRNA-239, elinkaari on huomattavasti pidentynyt, kun taas C. elegans -mutaatioissa, joissa on poistettu miRNA-71, miRNA-238 ja miRNA-246, tutkittavalla eläimellä havaitaan merkittävästi lyhyempi elinikä [ 175].
Ames-kääpiöhiiren pitkäikäisyys – johtuen lisääntyneestä insuliiniherkkyydestä, lisääntyneestä stressiresistenssistä ja vähentyneestä kasvaintiheydestä IGF-1-aktiivisuuden vähenemisen seurauksena – liittyi maksan miRNA-27a-suppression säätelyproteiineihin, ornitiinidekarboksylaasiin ja spermidiinisyntaasiin [176] .
Energiansaannin rajoittaminen muuttaa miRNA-ilmentymisprofiilia. Hiirillä, jotka saivat 70% normaalista energiasta 6 kuukauden ajan, kaloreiden rajoittaminen lisäsi miR-203:n ilmentymistä. Muutos kohdistuu kaveolin-1:n ja p63:n määriin, jotka vaikuttavat syöpäsolujen kasvuun ja invasiiviseen potentiaaliin [177]. Tutkijat päättelivät tästä, että kaloreiden rajoittaminen voi vähentää rintasyövän ilmaantuvuutta, etenemistä ja etäpesäkkeiden kehittymistä, mikä lisää odotettavissa olevia elinvuosia.
Kalorirajoitettujen hiirten aivot osoittivat miRNA-181a:n, miRNA-30e:n ja miRNA-34a:n vähentymistä, kun kaloreiden rajoittaminen oli jatkunut 8 kuukautta energiansaannin ollessa 60 % normaalista. Samalla BCL2-ilmentymisen havaittiin lisääntyvän ja BAX-ilmentymisen vähenevän, mikä vaikutti pienempiin kaspaasien 9 ja -3 aktiivisuuksiin. Kaspaasien 9 ja 3 aktiivisuuden heikkeneminen liittyy alentuneeseen apoptoosinopeuteen [178]. BAX- ja kaspaasi-3 -aktiivisuus lisääntyvät myös Alzheimerin ja Parkinsonin taudeissa [179-183].
6. Kalorirajoitusta jäljittelevät ruokavaliot
Koska pitkäaikainen kaloreiden saannin rajoittaminen on tarpeen koeolosuhteissa havaittujen myönteisten terveys- ja pitkäikäisyysvaikutusten aikaansaamiseksi, on etsitty vaihtoehtoja, jotka voisivat tuottaa kaloreiden rajoittamisen positiivisia vaikutuksia ilman ravinnon saantiin liittyviä rajoituksia.
Ihanteellisen ruokavalion tulisi:
(a) saada aikaan samanlaisia aineenvaihduta-, hormonaalisia ja fysiologisia vaikutuksia kuin kaloreiden rajoittaminen
(b) sen ei tulisi edellyttää merkittävää vähennystä pitkäaikaisessa ruoan saannissa
(c) sen pitäisi aktivoida energiansaannin rajoittamisen n kaltaiset stressivastereitit
(d) pidentää elinikää sekä vähentää tai viivästyttää ikään liittyvien sairauksien puhkeamista [184].
Tällaisen ruokavalion löytämiseksi kansallinen ikääntymisinstituutti perusti interventioiden testausohjelman testatakseen aineita, joiden ennustetaan pidentävän elinikää ja viivästyttävän sairauksia ja toimintahäiriöitä [185–189].

6.1. Kaloreiden rajoittaminen ja liikunta
Urosrottia suositaan eläinkokeissa joissa tutkitaan voiko liikunta yhdessä kaloreita rajoittavan ruokavalion kanssa toisintaa pelkästään kaloreita rajoittavan ruokavalion tuottamat edut. Kysymys on sikäli aiheellinen, etä tutkittavien rottien energiansaantia ei lisätä kompensoimaan lisääntyneen kulutuksen luomaa energiavajetta[ 180].
Eräissä tutkimuksissa on havaittu, että liikunnan ja kaloreiden rajoittamisen yhdistämisellä ei ole terveyttä edistäviä etuja, jotka ylittävät pelkällä kaloreiden rajoittamisella saavutetut edut [111, 190–192]. Näiden tutkimusten mukaan liikunta ei tuo lisäarvoa kaloreiden rajoittamista koituville fysiologisille hyödyille. Oksidatiivisen stressin tasoissa tai tulehdusta edistävien proteiinien pitoisuuksissa ei tapahtunut merkittävää muutosta liikkumaan päässeissä eläimissä, joiden energiansaantia oli laskettu 80% normaalista [191, 192]. Liikunnalla ei myöskään ollut vaikutusta eläimen maksimaaliseen elinaikaan [190].
Toisaalta, liikunnan ja vähäkalorisen ravinnon yhdistelmä pienensi hiljaista tulehdusta ilmentäviä CRP-tasoja enemmän kuin pelkkä kaloreiden rajoittaminen [193] ja pienensi sydänlihaksen nekroosin että sydänlihaksen iskemian kehittymisen riskiä [194, 195].
Useissa kaloreita rajoittavien ja liikuntaa lisäävien CE-tutkimusten ihmismalleissa on selvitetty 25 % kokonaiskaloripitoisuuden vähentämisen vaikutuksia terveyteen, kun 12,5% kaloreiden vähennyksestä tuli liikunnan lisäämästä energian kulutuksesta ja toinen 12,5% ravinnon pienemmästä energiapitoisuudesta.
Useimmissa tutkimuksissa paastoinsuliinin tasoissa, DNA-vaurioissa, lihasten mitokondrioiden geeniekspressioissa, triglyseriditasoissa ja maksan lipidipitoisuudessa ei havaittu merkittäviä eroja pelkän energian rajoittamisen ja energian rajoittamisen ja liikunnan yhdistämisen välillä [76, 196–198]. Poikkeuksena olivat kaksi tutkimusta, joissa raportoitiin sekä diastolisen verenpaineen että LDL-kolesterolin laskua edelleen, kun kaloreiden rajoittamisen yhdistämistä liikuntaan verrattiin pelkkään kaloreiden rajoittamiseen [198, 199].
Kaloreiden rajoittamisen ja liikunnan yhdistämisen on osoitettu lisäävän luun mineraalitiheyttä reisiluun kaulassa ja vähentävän tulehduksellista biomarkkeri sTNFR1:tä ylipainoisilla postmenopausaalisilla naisilla [200].
Suurin etu kaloreiden rajoittamisen yhdistämisestä liikuntaan verrattuna pelkästään kaloreiden rajoittamiseen on se, että ihmisen voi olla helpompaa noudattaa hoito-ohjelmaa, jossa kokonaisenergian lasku (ts. kalorien vähennys) jaetaan liikunnan lisäämän energian kulutuksen ja kalorien rajoitusten välillä [201].
6.2. Ruokavalion makroravinteiden rajoittaminen (DR)
Ruokavalion rajoittaminen (DR) viittaa proteiinin, rasvan ja hiilihydraattien välisten saantisuhteiden muuttamiseen joko vähentämällä tai vähentämättä kokonaiskalorien saantia. Hiilihydraattien ja rasvojen rajoituksista on saatu hyvin erilaisia tutkimustuloksia hyvin erilaisilla saantimäärillä. Tämän mukaan rasvan tai hiilihydraattien rajoittaminen ei vähennä reaktiivisten happilajien tuotantoa tai oksidaatioon perustuvia DNA-vaurioita [202–208].
Eläinmallissa proteiinin rajoittaminen näyttää olevan vaihtoehto kaloreiden rajoittamiselle. Proteiinin rajoittamisen raportoitiin lisäävän jyrsijöiden maksimaalista elinikää 20% [206]. DR-proteiinin elinikää pidentävät edut johtuivat metioniinirajoituksesta ruokavaliossa [209–215]. Esimerkiksi 40-prosenttisen metioniinirajoituksen on raportoitu vähentävän sekä mitokondrioiden reaktiivisten happilajien muodostumista että oksidatiivisia vaurioita mitokondrioiden DNA:ssa [216, 217].
Todisteet, jotka tukevat metioniinirajoituksen ja pidemmän eliniän välistä yhteyttä, sisältävät:
(a) käänteisen suhteen metioniinipitoisuuden ja nisäkkäiden maksimaalisen eliniän välillä [218]
(b) metioniini lisää LDL-kolesterolin hapettumista [219]
(c) lisääntynyt metioniinin saanti lisää plasman homokysteiinipitoisuuksia ja siten sydän- ja verisuonitautien ja kuolleisuuden riskiä [219].
On myös osoitettu, että kaikkien ravinnon aminohappojen rajoittaminen 40% metioniinia lukuun ottamatta ei vähennä reaktiivisten happilajien muodostumista tai oksidatiivisia vaurioita mitokondrioiden DNA:ssa [220]. Eräs ongelma voi siis olla liian runsas metioniinin saanti.
Yhteenvetona eläinkokeet viittaavat siihen, että noin puolet kaloreiden rajoittamisen eliniän pidentämisvaikutuksesta voidaan katsoa johtuvan metioniinirajoituksesta [206]. Siksi tutkimusten jatkaminen ihmisillä on perusteltua, koska metioniinin rajoittaminen on toteutettavissa ja hyvin siedetty [221].
6.3. Pätkäpaasto
Pätkäpaasto (ADF) vuorottelee 24 tunnin ad libitum -saannin jaksoja kalorien saannin osittaisella tai täydellisellä rajoittamisella. ADF ei välttämättä vähennä energian kokonaissaantia tai painoa, koska henkilöt voivat kompensoida alentuneen energiansaannin syömällä enemmän paasto-aikojen ulkopuolella [222, 223].
ADF pidensi koe-eläinten elinikää eläinkokeissa [223–225]. Jotkut tutkijat pitivät ADF:n vaikutusta elinajan pidentymiseen seurauksena aivoperäisen neurotrofisen tekijän samanaikaisen lisääntymisen kanssa [215].
ADF myös hillitsi tai esti ikään liittyvien sairausprosessien, kuten sydän- ja verisuonitautien, munuaissairauksien, syöpien ja diabeteksen kehittymistä [222, 223, 225–230].
Ihmiskokeissa on osoitettu, että pätkäpaasto on toteutettavissa, turvallinen ja hyvin siedetty ruokavalio myös ihmisillä [231]. ADF-ihmiskokeiden alustavia tuloksia [231–233] ei kuitenkaan voida verrata kaloreita rajoittaviin ihmiskokeisiin, koska ADF-kokeiden jaksot ovat olleet suhteellisen lyhyitä (muutamasta päivästä 20 viikkoon) verrattuna kalorirajoitus-kokeisiin (6 kuukaudesta 6 vuoteen) [74, 83, 85].
Kestoltaan jopa hyvin lyhyissä paastotutkimuksissa havaittiin joitain potentiaalisesti hyödyllisiä vaikutuksia, kuten: paastoinsuliinin lasku ilman eroja paastoglukoosissa [231] ja parantunut keuhkoputkien vaste lääkkeille [233].
On raportoitu, että normaalipainoisilla keski-ikäisillä koehenkilöillä, 2 kuukauden pituisen pätkäpaaston vaikutuksesta perifeerisen veren mononukleaariset solut tuottivat vähemmän tulehduksellisia sytokiineja [234].
Tämän katsauksen kirjoittamisen aikaan pätkäpaaston vaikutuksista veren lipideihin ja oksidatiiviselle stressille ominaisten biomarkkereiden tasoihin ei vielä ollut kovinkaan paljon tietoa.
6.4. Resveratroli
Resveratroli (RSV) on kasviperäinen mm. mustikoiden, karpaloiden, viinimarjojen ja punaviinin sisältämä polyfenoli, joka on eniten tutkittu kalorirajoitusta jäljittelevä aine.
Resveratrolin on osoitettu aktivoivan Sir2:n (SIRT1-homologi) [235] ja jäljittelemällä siten kaloreiden rajoittamisen etuja rajoittamatta kalorien saantia. Resveratroli on lisännyt hiivan, matojen, kärpästen ja kalojen elinikää [235–238].
Oletus, että Sir2:n aktivaatio suoralla sitoutumisella RSV:n kanssa vaikuttaisi elinajan pidentymiseen, on kuitenkin kyseenalaistettu useillaa organismeillaa tehdyissä kokeissa [239–248].
Resveratrolin tiedetään vaikuttavan laajasti nisäkässoluissa, kuten AMP-aktivoidun proteiinikinaasin (AMPK) aktivaatiossa. AMPK on mukana samoissa aineenvaihduntareiteissä kuin SIRT1, joka fosforyloi suoraan PGC-1α:n. [249, 250].
SIRT1 voi aktivoida kinaasin ylävirtaan AMPK:sta, mutta tämä reitti ei näytä olevan tarpeen resveratrolin AMPK-stimulaatiossa [251]. Äskettäin raportoitiin, että SIRT1 on välttämätön kohtuullisille resveratroliannoksille AMPK:n stimuloimiseksi ja mitokondrioiden toiminnan parantamiseksi in vitro ja in vivo [252]. Vaikka resveratroli-välitteisten kalorirajoituksen kaltaisten vaikutusten mekanismia ei täysin ymmärretä, näyttää siltä, että resveratroli tuottaa samanlaisen transkriptiovasteen kuin kaloreiden rajoittaminen [253]. Resveratrolia sisältävä ruunsaasti rasvaa sisältävän ruokavalio on tuottanut terveys- että pitkäikäisyyshyötyjä hiirikokeissa [249].
Resveratrolin käytön myönteiset vaikutukset lihavilla hiirillä olivat lisääntynyt insuliiniherkkyys, parantunut motorinen koordinaatio ja harmaakaihin esiintyvyyden väheneminen [253, 254]. Aikuisten hiirten elinajanodote ei noussut merkittävästi, kun resveratrolia lisättiin normaaliin ruokavalioon [254, 255]. Tämä havainto tarkoittaa, että resveratroli ei yksin tuota samoja hyötyjä kuin kaloreiden rajoittaminen [256]. Vuoden resveratroli-hoito lisäsi lepoaineenvaihdunnan nopeutta ja päivittäistä kokonaisenergiankulutusta. Resveratrolin pitkäaikainen käyttö on tehokasta ja turvallista [257, 258]. Kaloreiden rajoittaminen samassa eläinmallissa ja koeprotokollassa, vähensi päivittäistä kokonaisenergiankulutusta, mutta ei muuttanut lepoaineenvaihdunnan nopeutta [258].
Resveratrolin vaikutuksista ihmisiin on tehty vain muutama tutkimus, mutta tulokset ovat rohkaisevia. 0,1 mmol/l resveratrolin käyttö ihmisen mesenkymaalisten kantasolujen viljelmissä edistää solujen uudistumista estämällä solujen vanhenemista; suuremmilla pitoisuuksilla (5 mmol/l tai enemmän) resveratroli estää solujen uudistumista lisäämällä ikääntymisnopeutta, solujen kaksinkertaistumisaikaa ja S-vaiheen solusyklin pysäyttämistä [259].
Ihmisen peritoneaalisissa mesoteliaalisoluissa resveratroli viivästyttää replikatiivista vanhenemista mobilisoimalla antioksidatiivisia ja DNA-korjausmekanismeja solun tuma-antigeenin ilmentymisen lisääntymisellä, solujen lisääntyneellä fraktiolla solusyklin S-vaiheessa, lisääntyneellä solunjakautumisten määrällä, ikääntymiseen liittyvän β-galaktosidaasin vähentyneellä ilmentymisellä ja aktiivisuudella, mitokondrioiden säädellyn biogeneesin, superoksididismutaasin lisääntyneen aktiivisuuden ja vähentyneiden DNA-vaurioiden perusteella [260].
Resveratroli ja sen metaboliitit kertyvät ihmissoluihin in vivo kudosspesifisellä ja annosriippuvalla tavalla [261]. Kuuden viikon täydennysohjelma resveratrolilla tukahdutti tumatekijä kappa B:n (NF-kB) sitoutumisen, vähensi vapaiden happiradikaalien (ROS) muodostumista ja laski TNFα :n ja interleukiini-6:n (IL-6) tasoja yksitumaisissa soluissa. TNFα :n ja CRP: n pitoisuudet plasmassa laskivat myös merkittävästi. Resveratroli ei kuitenkaan vaikuttanut merkittävästi paasto-kolesterolin (kokonais-, LDL- ja HDL-pitoisuuksien), triglyseridien tai leptiinin pitoisuuksiin verrattuna terveiden plaseboa saaneiden henkilöiden kontrolliryhmään [262].
Runsasrasvainen ja hiilihydraattipitoinen ruokavalio aiheuttavat ja ylläpitävät tulehdusta ja oksidatiivista stressiä [263]. Terveillä ihmisillä, joiden ravinto sisältää runsaasti rasvaa ja hiilihydraatteja, resveratrolia ja muita rypäleiden sisältämien polyfenoleja sisältävä lisäaine lisäsi merkittävästi mRNA:n ilmentymistä NAD (P) H-dehydrogenaasi [kinoni] 1:n ja glutationin S-transferaasi-p1-geeneissä – mikä viittaa vahvaan antioksidanttivaikutukseen. Resveratroli lisäravinteena hillitsi aterian jälkeistä plasman endotoksiinia ja lipoproteiinia sitovan proteiinin pitoisuuden kasvua ja heikensi TLR-4:n, CD14:n, SOCS-3:n, IL-1β:n ja KEAP-1:n ilmentymistä [264].
Tutkimusten perusteella resveratroli vähentää runsaasti rasvaa ja runsaasti hiilihydraatteja sisältävän ravinnon aiheuttamia oksidatiivisia ja tulehduksellisia reaktioita, ja se voi vähentää ateroskleroosin ja diabeteksen riskiä [261].
Alustavien tulosten mukaan resveratroli parantaa myös glukoositoleranssia ja insuliiniherkkyyttä [265]. Parantunut insuliiniherkkyys johtui vähentyneestä oksidatiivisesta stressistä [265]. Syy-yhteys punaviinin ja rypälemehun kulutuksen ja sydän- ja verisuonitautien riskitekijöiden (verenkierron heikkeneminen, lisääntynyt oksidatiivinen stressi ja tulehdus) välillä on hyvin tunnettu [266–269].
Resveratroli säätelee eNOS:ta, mikä edistää typpioksidivälitteistä vasodilataatiota ja lisääntynyttä verenkiertoa [270–272]. Tämä vaikuttaa esimerkiksi erektioon. Resveratroli vaimentaa ihmisen verihiutaleiden hemostaasiin liittyvää aktivaatiota [273]. Lisääntynyt valtimoverenkierto mitattiin yhden resveratroli-boluksen jälkeen aivoissa ja käsivarressa [274, 275].
Lisääntynyt aivoverenkierto resveratroli-hoidon jälkeen ei kuitenkaan liittynyt lisääntyneeseen kognitiiviseen toimintaan [274]. Parantunut insuliiniresistenssi, valtimoverenkierto ja vähentynyt oksidatiivinen stressi ja tulehdus liittyvät resveratrolin lyhytaikaiseen käyttöön, mutta pitkäin aikavälin vaikutuksista ihmisiin ei ole tietoja [261]. Yhteenvetona voidaan todeta, että lisätutkimuksia tarvitaan resvetroli-välitteisten vaikutusten biokemiallisten reittien selventämiseksi ja sen pitkäaikaisten vaikutusten selvittämiseksi ihmisillä [276].
6.5. Rapamysiini
Rapamysiini (RAP) on antibiootti ja TOR:n (rapamysiinikohde) signaloinnin estäjä soluissa, joilla on tunnettuja immunosuppressiivisia ja antiproliferatiivisia vaikutuksia [277].
TOR on solujen ravinteiden signaloinnin välittäjä, ja sen uskotaan vaikuttavan ikääntymiseen ja kalorirajoitus-vasteeseen (ks. Kohta 6.3). Kun rapamysiiniä annettiin hiirille noin 20 kuukauden iässä, uros- ja naaraspuolisten hiirten keskimääräinen elinaika lisääntyi merkittävästi, noin 10%.
Rapamysiinin vaikutuksen voidaa ainakin osittain välittää kalorirajoitus-vasteesta riippumattomilla biokemiallisilla reiteillä [117]. Useiden rapamysiini-aktivoitujen ikääntymistä hidastavien biokemiallisten reittien olemassaolo on havaittu myös kärpäsissä.
Mekanismi tälle rapamysiinien elinaikaa lisäävien vaikutusten taustalla johtuu TOR-reitin TORC1-haarasta, autofagian ja translaation muutoksilla. Rapamysiini voi kuitenkin vaikuttaa suotuisasti elinaikaan kaloreiden rajoittamisesta riippumatta, mikä viittaa lisämekanismeihin eliniän pidentämiseksi [278].
Rapamysiini esti eläintutkimuksissa ikään liittyvää painonnousua, laski ikääntymisnopeutta, pidensi elinikää ja viivästytti spontaania syöpää [279]. Rapamysiinillä hoidetut aikuiset hiiret suoriutuivat huomattavasti paremmin spatiaalista oppimista ja muistia mittaavista tehtävistä, kuin saman ikäiset verrokit. Rapamysiini ei kuitenkaan parantanut kognitiota aikuisilla hiirillä, joilla oli ennestään, iästä riippuva oppimis. ja muistivaje. Rapamysiinivälitteinen oppimisen ja muistin paraneminen liittyi IL-1β-tasojen laskuun ja NMDA-signaloinnin lisääntymiseen. [280]. Koska rapamysiiniä käytetään immunosuppressiivisena aineena, sen merkitystä ihmisten pitkäikäisyydelle ei ole vielä vahvistettu [117].
7. Ruokavalio ja ikääntyvä väestö
Tärkeä väestörakenteen kehityssuuntaus kehittyneissä maissa on yli 65-vuotiaiden väestön prosentuaalisen osuuden asteittainen kasvu ja työikäisen väestön samanaikainen väheneminen.
Tämän demografisen suuntauksen terveysvaikutukset ovat siirtyminen akuuteista kroonisiin ja ikään liittyviin sairauksiin (esim. Alzheimerin tauti, osteoporoosi, sydän- ja verisuonitaudit ja syöpä), lisääntyvät terveyskustannukset ja kasvava taloudellinen taakka yhteiskunnalle ja yksilölle [281– 283].
Siksi kaikilla toimenpiteillä, jotka voivat viivästyttää kroonisten ja ikään liittyvien sairauksien etenemistä, voi olla merkittävä vaikutus paitsi yksilön elämänlaatuun myös yhteiskunnan kykyyn selviytyä ikääntymisen terveydellisistä ja taloudellisista seurauksista.
On olemassa jatkuvasti lisääntyvää tutkimusnäyttöä, jonka mukaan ravinnon energiapitoisuuden vähentäminen, pätkäpaasto ja ketogeeninen ruokavalio parantavat useimpia terveysmarkkereita verenpaineesta tulehdustekijöihin ja verensokerista insuliinipitoisuuteen ja lipiditasoihin.
Tutkimukset viittaavat siihen, että kaloreiden rajoittaminen voi vähentää merkittävästi ikään liittyvien muutosten määrää ihmisillä [73–93]. Poikkeuksellisen pitkäikäisillä ihmisillä tehdyt tutkimukset viittaavat siihen, että pitkäikäisyys ja ikään liittyvien sairauksien vähäinen esiintyvyys suvussa mahdollistavat huomattavasti pidemmän eliniän jopa silloin, kun tutkittavat olivat lihavia, tupakoivia tai eivät harrasta säännöllistä liikuntaa. Ihmisten poikkeuksellinen pitkäikäisyys voi olla enemmän riippuvainen genetiikasta kuin elämäntavasta [284–286].
8. Päätelmä
Kalorirajoitus tai kalorirajoitusta jäljittelevät ruokailutottumukset aiheuttavat koordinoituja adaptiivisia stressivasteita solujen ja koko organismin tasolla moduloimalla adiponektiinin, insuliini / IGF1, AMPK, mTOR, FOXO, p53 ja sirtuiinien signalointireittejä [287].
Sirtuiineilla voi olla tärkeä rooli epigeneettisten ja geneettisten reittien välisessä vuorovaikutuksessa [132]. Näiden adaptiivisten stressivasteiden aktivaatio voi estää apoptoosin alkamisen sisäisellä reitillä [288]. Lisäksi se voi stimuloida autofagiaa tarjoamaan substraatteja energiantuotannolle ja anabolisille prosesseille, jotka liittyvät solujen uudistumiseen ja antioksidanttien ja lämpöshokkiproteiinien synteesiin [287].
Suuri joukko kokeellisia todisteita osoittaa, että näiden mukautuvien stressivasteiden kokonaisvaikutuksena on lisääntynyt vastustuskyky stressille, mikä viivästyttää ikään liittyviä muutoksia ja edistää pitkäikäisyyttä.
Tämä on pitkä artikkeli. Pyydän anteeksi kirjoitus- ja käännösvirheitä. Artikkeli on vertaisarvioitu ja tieteellisessä julkaisussa julkaistu, joten molekyylibiologiset mekanismit ovat uskoakseni käännösvirheitä paitsi oikein. Ruokavalioiden suhteen juttu ei ole täysin ajan tasalla. Tieto ketogeenisen ruokavalion, paaston ja pätkäpaaston vastaavista molekyylibiologisista hyödyistä on lisääntynyt kuluneiden 10 vuoden aikana.
Lyhenteet
| 4E-BP1: | Eukaryotic translation initiation factor 4E binding protein 1 |
| ADF: | Alternate day fasting |
| AGC: | Acronym of the protein kinase A, G, and C families |
| AKT: | Serine-threonine-specific proteinkinase also known as protein kinase B (PKB) |
| AMP: | Adenosine monophosphate |
| AMPK: | 5′ adenosine monophosphate-activated protein kinase |
| ATP: | Adenosine-5′-triphosphate |
| ATP10A: | Probable phospholipid-transporting ATPase VA also known as ATPase class V type 10A or aminophospholipid translocase VA gene |
| Aβ: | Amyloid beta |
| B12 vitamin: | Cobalamin |
| BAX: | Bcl-2 associated X protein |
| BCL-XL: | B-cell lymphoma-extra large, a transmembrane mitochondrial protein |
| CALERIE: | Comprehensive Assessment of Long-Term Effects of Reducing Calorie Intake |
| CD14: | Cluster of differentiation 14 protein also known as CD14 protein |
| CE: | Exercise in combination with CR |
| CHD: | Coronary heart disease |
| CpG dinucleotide: | Cytosine-phosphate-guanine dinucleotide |
| CR: | Caloric restriction or calorie restriction diet |
| CRM: | Calorie restriction mimetic |
| CRP: | C-reactive protein |
| CRS: | Caloric Restriction Society |
| DNA: | Deoxyribonucleic acid |
| DNMT: | DNA methyltransferase |
| DR: | Dietary restriction |
| E2F-1: | Transcription factor E2F1 protein |
| EGCG: | Epigallocatechin-3-gallate |
| eNOS: | Endothelial nitric oxide synthase |
| FOXO: | O subclass of the forkhead family of transcription factors; known FOXO family members are FOXO1, FOXO3, FOXO4 and FOXO6 |
| GLUT4: | Glucose transporter 4 |
| GTP: | Guanosine-5′-triphosphate |
| GTPase: | Enzyme that hydrolyses GTP |
| HAT(s): | Histone acetlytransferase(s) |
| HDAC(s): | Histone deacetylase(s) |
| HDAC(s)s: | Histone deacetylase(s) |
| HDL: | High-density lipoprotein |
| HDM(s): | Histone demethylase(s) |
| hmdC: | 5-hyd0072oxymethyl-2′-deoxycytidine |
| HNF4α: | Hepatocyte nuclear factor 4 α also known as nuclear receptor subfamily 2, group A, member 1 |
| HMT(s): | Histone methyltransferase(s) |
| HNF4α: | Hepatocyte nuclear factor 4α |
| HRV: | Heart-rate-variability |
| : | Gene encoding human telomerase reverse transcriptase a catalytic subunit of the enzymetelomerase |
| IEE: | Increased energy expenditure |
| IGF1: | Insulin-like growth factor 1 also known as somatomedin C |
| IL-1β: | Human interleukin 1β |
| c-KIT: | Proto-oncogene c-Kit also known as mast/stem cell growth factor receptor, also known as tyrosine-protein kinase Kit or CD117 |
| IRS: | Insulin receptor substrate |
| KEAP-1: | Kelch-like ECH-associated protein 1 |
| Ku70: | Protein encoded in humans by the gene |
| LBK1: | Tumor suppressor kinase enzyme that activates AMPK |
| LDL: | Low-density lipoprotein |
| miRNA(s): | microRNA(s) |
| mRNA: | Messenger RNA |
| mSin1: | Mammalian stress-activated protein kinase-interacting protein |
| MTH: | Mitochondrion, mitochondrial |
| mTOR: | Mammalian target of rapamycin |
| mTORC1: | Mammalian target of rapamycin complex 1 |
| mTORC2: | Mammalian target of rapamycin complex 2 |
| Nicotinamide adenine dinucleotide | |
| NADH: | NADH dehydrogenase |
| NF- |
| B: | nuclear factor kappa B |
| NIP7: | 60S ribosome subunit biogenesis protein NIP7 homolog |
| NMDA: | N-Methyl-D-aspartic acid or N-Methyl-D-aspartate |
| : | Gene encoding the tumor suppressor protein cyclin-dependent kinase inhibitor 2A or CDKN2A or multiple tumor suppressor 1 (MTS-1) |
| PDPK1: | 3-phosphoinositide-dependent protein kinase-1 |
| PGC1-α: | Peroxisome proliferator-activated receptor G co-activator 1α |
| p53: | Tumor suppressor protein p53 also known as tumor protein 53 |
| p47phox: | Subunit of NADPH oxidase, that has to be phosphorilated for the activation of NADPH oxidase |
| PKA: | Protein kinase A |
| PKC: | Protein kinase C |
| PKG: | Protein kinase G, or cGMP-dependent protein kinase |
| PtdIns-3K: | Phosphatidylinositol 3-kinase |
| RAP: | Rapamycin |
| RAPTOR: | Regulatory-associated protein of mTOR |
| RHEB: | RAS homolog enriched in brain protein, binds GTP |
| RNA: | Ribonucleic acid |
| ROS: | Reactive oxygen species |
| RSV: | Resveratrol |
| RAS: | Protein superfamily of small GTPases |
| RTG1: | Retrograde regulation protein 1 |
| RUNX3: | Gene encoding runt-related transcription factor 3 |
| S6 K1: | Ribosomal protein S6 kinase |
| -1 | |
| SGK1: | Serum-and glucocorticoid-regulated kinase; a serine/threonine protein kinase |
| SIRT1: | NAD-dependent-deacetylase sirtuin1 also known as silent mating type information regulation 2 homolog 1 |
| SOCS-3: | Suppressor of cytokine signaling 3 |
| sTNRF1: | Soluble tumor necrosis factor receptor 1 |
| TLR-4: | Toll-like receptor 4 |
| TNFα: | Tumor necrosis factor α |
| TOR: | Target of rapamycin |
| TSC1: | Tuberous sclerosis protein 1 also known as hamartin |
| TSC2: | Tuberous sclerosis protein 2 also known as tuberin |
| VDAC1: | Voltage-dependent anion-selective channel protein 1 |
| TIG1: | Tazarotene-induced gene-1 |
| WT1: | Gene encoding Wilms tumor protein |
| YY1: | Transcriptional repressor protein YY1. |

References
- J. A. Mckay and J. C. Mathers, “Diet induced epigenetic changes and their implications for health,” Acta Physiologica, vol. 202, no. 2, pp. 103–118, 2011. View at: Publisher Site | Google Scholar
- “Diet, nutrition and the prevention of chronic diseases,” World Health Organization Technical Report Series, vol. 916, no. 1–8, pp. 1–149, 2003. View at: Google Scholar
- D. J. P. Barker and C. Osmond, “Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales,” The Lancet, vol. 1, no. 8489, pp. 1077–1081, 1986. View at: Google Scholar
- O. A. Kensara, S. A. Wootton, D. I. Phillips, M. Patel, A. A. Jackson, and M. Elia, “Fetal programming of body composition: relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen,” The American Journal of Clinical Nutrition, vol. 82, no. 5, pp. 980–987, 2005. View at: Google Scholar
- C. Osmond, D. J. P. Barker, P. D. Winter, C. H. D. Fall, and S. J. Simmonds, “Early growth and death from cardiovascular disease in women,” British Medical Journal, vol. 307, no. 6918, pp. 1519–1524, 1993. View at: Google Scholar
- C. N. Hales and D. J. P. Barker, “Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis,” Diabetologia, vol. 35, no. 7, pp. 595–601, 1992. View at: Publisher Site | Google Scholar
- C. Cooper, C. Fall, P. Egger, R. Hobbs, R. Eastell, and D. Barker, “Growth in infancy and bone mass in later life,” Annals of the Rheumatic Diseases, vol. 56, no. 1, pp. 17–21, 1997. View at: Google Scholar
- S. E. Ozanne and C. N. Hales, “Lifespan: catch-up growth and obesity in male mice,” Nature, vol. 427, no. 6973, pp. 411–412, 2004. View at: Google Scholar
- V. M. Vehaskari, “Prenatal programming of kidney disease,” Current Opinion in Pediatrics, vol. 22, no. 2, pp. 176–182, 2010. View at: Publisher Site | Google Scholar
- A. Gabory, L. Attig, and C. Junien, “Sexual dimorphism in environmental epigenetic programming,” Molecular and Cellular Endocrinology, vol. 304, no. 1-2, pp. 8–18, 2009. View at: Publisher Site | Google Scholar
- A. Bird, “DNA methylation patterns and epigenetic memory,” Genes & Development, vol. 16, no. 1, pp. 6–21, 2002. View at: Publisher Site | Google Scholar
- H. Wu, J. Tao, and Y. E. Sun, “Regulation and function of mammalian DNA methylation patterns: a genomic perspective,” Briefings in Functional Genomics, vol. 11, no. 3, pp. 240–250, 2012. View at: Google Scholar
- X. Zou, W. Ma, I. A. Solov’yov, C. Chipot, and K. Schulten, “Recognition of methylated DNA through methyl-CpG binding domain proteins,” Nucleic Acids Research, vol. 40, no. 6, pp. 2747–2758, 2012. View at: Google Scholar
- K. S. Crider, T. P. Yang, R. J. Berry, and L. B. Bailey, “Folate and DNA methylation: a review of molecular mechanisms and the evidence for folate’s role,” Advances in Nutrition, vol. 3, no. 1, pp. 21–38, 2012. View at: Google Scholar
- T. A. Rauch, X. Zhong, X. Wu et al., “High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer,” Proceedings of the National Academy of Sciences of the United States of America, vol. 105, no. 1, pp. 252–257, 2008. View at: Publisher Site | Google Scholar
- M. Ehrlich, “DNA hypomethylation in cancer cells,” Epigenomics, vol. 1, pp. 239–259, 2009. View at: Google Scholar
- C. D. Davis, E. O. Uthus, and J. W. Finley, “Dietary selenium and arsenic affect DNA methylation in vitro in Caco-2 cells and in vivo in rat liver and colon,” The Journal of Nutrition, vol. 130, no. 12, pp. 2903–2909, 2000. View at: Google Scholar
- H. Zeng, L. Yan, W. H. Cheng, and E. O. Uthus, “Dietary selenomethionine increases exon-specific DNA methylation of the p53 gene in rat liver and colon mucosa,” The Journal of Nutrition, vol. 141, no. 8, pp. 1464–1468, 2011. View at: Publisher Site | Google Scholar
- E. M. E. Van Straten, V. W. Bloks, N. C. A. Huijkman et al., “The liver X-receptor gene promoter is hypermethylated in a mouse model of prenatal protein restriction,” American Journal of Physiology, vol. 298, no. 2, pp. R275–R282, 2010. View at: Publisher Site | Google Scholar
- A. J. Bannister and T. Kouzarides, “Regulation of chromatin by histone modifications,” Cell Research, vol. 21, no. 3, pp. 381–395, 2011. View at: Publisher Site | Google Scholar
- A. Link, F. Balaguer, and A. Goel, “Cancer chemoprevention by dietary polyphenols: promising role for epigenetics,” Biochemical Pharmacology, vol. 80, no. 12, pp. 1771–1792, 2010. View at: Publisher Site | Google Scholar
- X. Cheng and R. M. Blumenthal, “Coordinated chromatin control: structural and functional linkage of DNA and histone methylation,” Biochemistry, vol. 49, no. 14, pp. 2999–3008, 2010. View at: Publisher Site | Google Scholar
- Y. Tan, B. Zhang, T. Wu et al., “Transcriptional inhibiton of Hoxd4 expression by miRNA-10a in human breast cancer cells,” BMC Molecular Biology, vol. 10, article 12, 2009. View at: Publisher Site | Google Scholar
- P. G. Hawkins and K. V. Morris, “RNA and transcriptional modulation of gene expression,” Cell Cycle, vol. 7, no. 5, pp. 602–607, 2008. View at: Google Scholar
- D. P. Bartel, “MicroRNAs: target recognition and regulatory functions,” Cell, vol. 136, no. 2, pp. 215–233, 2009. View at: Publisher Site | Google Scholar
- B. Kusenda, M. Mraz, J. Mayer, and S. Pospisilova, “MicroRNA biogenesis, functionality and cancer relevance,” Biomedical papers of the Medical Faculty of the University Palacký, Olomouc, Czechoslovakia, vol. 150, no. 2, pp. 205–215, 2006. View at: Google Scholar
- I. Bentwich, A. Avniel, Y. Karov et al., “Identification of hundreds of conserved and nonconserved human microRNAs,” Nature Genetics, vol. 37, no. 7, pp. 766–770, 2005. View at: Publisher Site | Google Scholar
- B. P. Lewis, C. B. Burge, and D. P. Bartel, “Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets,” Cell, vol. 120, no. 1, pp. 15–20, 2005. View at: Publisher Site | Google Scholar
- R. C. Friedman, K. K. H. Farh, C. B. Burge, and D. P. Bartel, “Most mammalian mRNAs are conserved targets of microRNAs,” Genome Research, vol. 19, no. 1, pp. 92–105, 2009. View at: Publisher Site | Google Scholar
- L. P. Lim, N. C. Lau, E. G. Weinstein et al., “The microRNAs of Caenorhabditis elegans,” Genes & Development, vol. 17, no. 8, pp. 991–1008, 2003. View at: Publisher Site | Google Scholar
- A. Esquela-Kerscher and F. J. Slack, “Oncomirs—microRNAs with a role in cancer,” Nature Reviews Cancer, vol. 6, no. 4, pp. 259–269, 2006. View at: Publisher Site | Google Scholar
- J. C. Mathers, G. Strathdee, and C. L. Relton, “Induction of epigenetic alterations by dietary and other environmental factors,” Advances in Genetics, vol. 71, pp. 4–39, 2010. View at: Publisher Site | Google Scholar
- D. Milenkovic, C. Deval, E. Gouranton et al., “Modulation of miRNA expression by dietary polyphenols in apoE deficient mice: a new mechanism of the action of polyphenols,” PLoS One, vol. 7, no. 1, Article ID e29837, 2012. View at: Google Scholar
- Q. Sun, R. Cong, H. Yan et al., “Genistein inhibits growth of human uveal melanoma cells and affects microRNA-27a and target gene expression,” Oncology Reports, vol. 22, no. 3, pp. 563–567, 2009. View at: Publisher Site | Google Scholar
- M. Sun, Z. Estrov, Y. Ji, K. R. Coombes, D. H. Harris, and R. Kurzrock, “Curcumin (diferuloylmethane) alters the expression profiles of microRNAs in human pancreatic cancer cells,” Molecular Cancer Therapeutics, vol. 7, no. 3, pp. 464–473, 2008. View at: Publisher Site | Google Scholar
- J. Yang, Y. Cao, J. Sun, and Y. Zhang, “Curcumin reduces the expression of Bcl-2 by upregulating miR-15a and miR-16 in MCF-7 cells,” Medical Oncology, vol. 27, no. 4, pp. 1114–1118, 2010. View at: Publisher Site | Google Scholar
- S. Careccia, S. Mainardi, A. Pelosi et al., “A restricted signature of miRNAs distinguishes APL blasts from normal promyelocytes,” Oncogene, vol. 28, no. 45, pp. 4034–4040, 2009. View at: Publisher Site | Google Scholar
- F. U. Weiss, I. J. Marques, J. M. Woltering et al., “Retinoic acid receptor antagonists inhibit miR-10a expression and block metastatic behavior of pancreatic cancer,” Gastroenterology, vol. 137, no. 6, pp. 2136–2145, 2009. View at: Publisher Site | Google Scholar
- L. A. Davidson, N. Wang, M. S. Shah, J. R. Lupton, I. Ivanov, and R. S. Chapkin, “n-3 Polyunsaturated fatty acids modulate carcinogen-directed non-coding microRNA signatures in rat colon,” Carcinogenesis, vol. 30, no. 12, pp. 2077–2084, 2009. View at: Publisher Site | Google Scholar
- T. Melkamu, X. Zhang, J. Tan, Y. Zeng, and F. Kassie, “Alteration of microRNA expression in vinyl carbamate-induced mouse lung tumors and modulation by the chemopreventive agent indole-3-carbinol,” Carcinogenesis, vol. 31, no. 2, Article ID bgp208, pp. 252–258, 2010. View at: Publisher Site | Google Scholar
- C. J. Marsit, K. Eddy, and K. T. Kelsey, “MicroRNA responses to cellular stress,” Cancer Research, vol. 66, no. 22, pp. 10843–10848, 2006. View at: Publisher Site | Google Scholar
- H. Kutay, S. Bai, J. Datta et al., “Downregulation of miR-122 in the rodent and human hepatocellular carcinomas,” Journal of Cellular Biochemistry, vol. 99, no. 3, pp. 671–678, 2006. View at: Publisher Site | Google Scholar
- R. Loewith and M. N. Hall, “Target of rapamycin (TOR) in nutrient signaling and growth control,” Genetics, vol. 189, no. 4, pp. 1177–1201, 2011. View at: Google Scholar
- M. Laplante and D. M. Sabatini, “mTOR signaling in growth control and disease,” Cell, vol. 149, no. 2, pp. 274–293, 2012. View at: Google Scholar
- W. L. Yen and D. J. Klionsky, “How to live long and prosper: autophagy, mitochondria, and aging,” Physiology, vol. 23, no. 5, pp. 248–262, 2008. View at: Publisher Site | Google Scholar
- L. R. Pearce, D. Komander, and D. R. Alessi, “The nuts and bolts of AGC protein kinases,” Nature Reviews, vol. 11, no. 1, pp. 9–22, 2010. View at: Publisher Site | Google Scholar
- Y. Sancak, C. C. Thoreen, T. R. Peterson et al., “PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase,” Molecular Cell, vol. 25, no. 6, pp. 903–915, 2007. View at: Publisher Site | Google Scholar
- C. A. Easley IV, A. Ben-Yehudah, C. J. Redinger et al., “MTOR-mediated activation of p70 S6K induces differentiation of pluripotent human embryonic stem cells,” Cellular Reprogramming, vol. 12, no. 3, pp. 263–273, 2010. View at: Publisher Site | Google Scholar
- L. A. Julien, A. Carriere, J. Moreau, and P. P. Roux, “mTORC1-activated S6K1 phosphorylates rictor on threonine 1135 and regulates mTORC2 signaling,” Molecular and Cellular Biology, vol. 30, no. 4, pp. 908–921, 2010. View at: Publisher Site | Google Scholar
- A. Y. Choo, S. O. Yoon, S. G. Kim, P. P. Roux, and J. Blenis, “Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation,” Proceedings of the National Academy of Sciences of the United States of America, vol. 105, no. 45, pp. 17414–17419, 2008. View at: Publisher Site | Google Scholar
- D. Zhang, R. Contu, M. V. G. Latronico et al., “MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice,” The Journal of Clinical Investigation, vol. 120, no. 8, pp. 2805–2816, 2010. View at: Publisher Site | Google Scholar
- R. J. O. Dowling, I. Topisirovic, T. Alain et al., “mTORCI-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs,” Science, vol. 328, no. 5982, pp. 1172–1176, 2010. View at: Publisher Site | Google Scholar
- K. G. Foster, H. A. Acosta-Jaquez, Y. Romeo et al., “Regulation of mTOR complex 1 (mTORC1) by raptor Ser863 and multisite phosphorylation,” The Journal of Biological Chemistry, vol. 285, no. 1, pp. 80–94, 2010. View at: Publisher Site | Google Scholar
- D. Kwak, S. Choi, H. Jeong et al., “Osmotic stress regulates mammalian target of rapamycin(mTOR) complex 1 via c-Jun N-terminal Kinase (JNK)-mediated Raptor protein phosphorylation,” The Journal of Biological Chemistry, vol. 287, no. 22, pp. 18398–18407, 2012. View at: Google Scholar
- T. Sato, A. Nakashima, L. Guo, and F. Tamanoi, “Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein,” The Journal of Biological Chemistry, vol. 284, no. 19, pp. 12783–12791, 2009. View at: Publisher Site | Google Scholar
- D. M. Gwinn, D. B. Shackelford, D. F. Egan et al., “AMPK phosphorylation of raptor mediates a metabolic checkpoint,” Molecular Cell, vol. 30, no. 2, pp. 214–226, 2008. View at: Publisher Site | Google Scholar
- B. Magnuson, B. Ekim, and D. C. Fingar, “Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks,” The Biochemical Journal, vol. 441, no. 1, pp. 1–21, 2012. View at: Google Scholar
- B. C. Melnik, “Excessive Leucine-mTORC1-signalling of cow milk-based infant formula: the missing link to understand early childhood obesity,” Journal of Obesity, vol. 2012, Article ID 197653, 2012. View at: Google Scholar
- A. K. A. DeHart, J. D. Schnell, D. A. Allen, J. Y. Tsai, and L. Hicke, “Receptor internalization in yeast requires the Tor2-Rho1 signaling pathway,” Molecular Biology of the Cell, vol. 14, no. 11, pp. 4676–4684, 2003. View at: Publisher Site | Google Scholar
- T. Powers, S. Aronova, and B. Niles, “TORC2 and sphingolipid biosynthesis and signaling. lessons from budding yeast,” The Enzymes, vol. 27, pp. 177–197, 2010. View at: Publisher Site | Google Scholar
- V. Zinzalla, D. Stracka, W. Oppliger, and M. N. Hall, “Activation of mTORC2 by association with the ribosome,” Cell, vol. 144, no. 5, pp. 757–768, 2011. View at: Publisher Site | Google Scholar
- A. Hagiwara, M. Cornu, N. Cybulski et al., “Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c,” Cell Metabolism, vol. 15, no. 5, pp. 725–738, 2012. View at: Google Scholar
- N. Cybulski and M. N. Hall, “TOR complex 2: a signaling pathway of its own,” Trends in Biochemical Sciences, vol. 34, no. 12, pp. 620–627, 2009. View at: Publisher Site | Google Scholar
- C. A. Sparks and D. A. Guertin, “Targeting mTOR: prospects for mTOR complex 2 inhibitors in cancer therapy,” Oncogene, vol. 29, no. 26, pp. 3733–3744, 2010. View at: Publisher Site | Google Scholar
- N. Ikai, N. Nakazawa, T. Hayashi, and M. Yanagida, “The reverse, but coordinated, roles of Tor2 (TORC1) and Tor1 (TORC2) kinases for growth, cell cycle and separase-mediated mitosis in Schizosaccharomyces pombe,” Open Biology, vol. 1, 2011. View at: Publisher Site | Google Scholar
- M. A. Frias, C. C. Thoreen, J. D. Jaffe et al., “mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s,” Current Biology, vol. 16, no. 18, pp. 1865–1870, 2006. View at: Publisher Site | Google Scholar
- C. M. McCay, M. F. Crowell, and L. A. Maynard, “The effect of retarded growth upon the Length of life span and upon the ultimate body size,” The Journal of Nutrition, vol. 10, no. 1, pp. 63–79, 1935. View at: Google Scholar
- R. J. Colman, R. M. Anderson, S. C. Johnson et al., “Caloric restriction delays disease onset and mortality in rhesus monkeys,” Science, vol. 325, no. 5937, pp. 201–204, 2009. View at: Publisher Site | Google Scholar
- R. Anderson and R. Weindruch, “Metabolic reprogramming in dietary restriction,” Interdisciplinary Topics in Gerontology, vol. 35, pp. 18–38, 2007. View at: Publisher Site | Google Scholar
- B. K. Kennedy, K. K. Steffen, and M. Kaeberlein, “Ruminations on dietary restriction and aging,” Cellular and Molecular Life Sciences, vol. 64, no. 11, pp. 1323–1328, 2007. View at: Publisher Site | Google Scholar
- M. D. W. Piper and A. Bartke, “Diet and aging,” Cell Metabolism, vol. 8, no. 2, pp. 99–104, 2008. View at: Publisher Site | Google Scholar
- G. S. Roth, D. K. Ingram, and M. A. Lane, “Caloric restriction in primates and relevance to humans,” Annals of the New York Academy of Sciences, vol. 928, pp. 305–315, 2001. View at: Google Scholar
- R. L. Walford, D. Mock, R. Verdery, and T. MacCallum, “Calorie restriction in biosphere 2: alterations in physiologic, hematologic, hormonal, and biochemical parameters in humans restricted for a 2-year period,” The Journals of Gerontology A, vol. 57, no. 6, pp. B211–B224, 2002. View at: Google Scholar
- L. Fontana, T. E. Meyer, S. Klein, and J. O. Holloszy, “Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans,” Proceedings of the National Academy of Sciences of the United States of America, vol. 101, no. 17, pp. 6659–6663, 2004. View at: Publisher Site | Google Scholar
- V. Tsagareli, M. Noakes, and R. J. Norman, “Effect of a very-low-calorie diet on in vitro fertilization outcomes,” Fertility and Sterility, vol. 86, no. 1, pp. 227–229, 2006. View at: Publisher Site | Google Scholar
- L. K. Heilbronn, L. De Jonge, M. I. Frisard et al., “Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial,” Journal of the American Medical Association, vol. 295, no. 13, pp. 1539–1548, 2006. View at: Publisher Site | Google Scholar
- S. B. Racette, E. P. Weiss, D. T. Villareal et al., “One year of caloric restriction in humans: feasibility and effects on body composition and abdominal adipose tissue,” The Journals of Gerontology A, vol. 61, no. 9, pp. 943–950, 2006. View at: Google Scholar
- T. E. Meyer, S. J. Kovács, A. A. Ehsani, S. Klein, J. O. Holloszy, and L. Fontana, “Long-term caloric restriction ameliorates the decline in diastolic function in humans,” Journal of the American College of Cardiology, vol. 47, no. 2, pp. 398–402, 2006. View at: Publisher Site | Google Scholar
- L. Fontana, S. Klein, J. O. Holloszy, and B. N. Premachandra, “Effect of long-term calorie restriction with adequate protein and micronutrients on thyroid hormones,” The Journal of Clinical Endocrinology & Metabolism, vol. 91, no. 8, pp. 3232–3235, 2006. View at: Publisher Site | Google Scholar
- L. Fontana and S. Klein, “Aging, adiposity, and calorie restriction,” Journal of the American Medical Association, vol. 297, no. 9, pp. 986–994, 2007. View at: Publisher Site | Google Scholar
- J. O. Holloszy and L. Fontana, “Caloric restriction in humans,” Experimental Gerontology, vol. 42, no. 8, pp. 709–712, 2007. View at: Publisher Site | Google Scholar
- B. J. Willcox, D. C. Willcox, H. Todoriki et al., “Caloric restriction, the traditional okinawan diet, and healthy aging: the diet of the world’s longest-lived people and its potential impact on morbidity and life span,” Annals of the New York Academy of Sciences, vol. 1114, pp. 434–455, 2007. View at: Publisher Site | Google Scholar
- L. Fontana, D. T. Villareal, E. P. Weiss et al., “Calorie restriction or exercise: effects on coronary heart disease risk factors. A randomized, controlled trial,” American Journal of Physiology, vol. 293, no. 1, pp. E197–E202, 2007. View at: Publisher Site | Google Scholar
- J. O. Holloszy and L. Fontana, “Caloric restriction in humans,” Experimental Gerontology, vol. 42, no. 8, pp. 709–712, 2007. View at: Publisher Site | Google Scholar
- T. Hofer, L. Fontana, S. D. Anton et al., “Long-term effects of caloric restriction or exercise on DNA and RNA oxidation levels in white blood cells and urine in humans,” Rejuvenation Research, vol. 11, no. 4, pp. 793–799, 2008. View at: Publisher Site | Google Scholar
- L. M. Redman, J. Rood, S. D. Anton, C. Champagne, S. R. Smith, and E. Ravussin, “Calorie restriction and bone health in young, overweight individuals,” Archives of Internal Medicine, vol. 168, no. 17, pp. 1859–1866, 2008. View at: Publisher Site | Google Scholar
- C. Cruzen and R. J. Colman, “Effects of caloric restriction on cardiovascular aging in non-human primates and humans,” Clinics in Geriatric Medicine, vol. 25, no. 4, pp. 733–743, 2009. View at: Publisher Site | Google Scholar
- R. Cangemi, A. J. Friedmann, J. O. Holloszy, and L. Fontana, “Long-term effects of calorie restriction on serum sex-hormone concentrations in men,” Aging Cell, vol. 9, no. 2, pp. 236–242, 2010. View at: Publisher Site | Google Scholar
- J. F. Trepanowski and R. J. Bloomer, “The impact of religious fasting on human health,” Nutrition Journal, vol. 9, no. 1, article 57, 2010. View at: Publisher Site | Google Scholar
- A. Soare, R. Cangemi, D. Omodei, J. O. Holloszy, and L. Fontana, “Long-term calorie restriction, but not endurance exercise, lowers core body temperature in humans,” Aging, vol. 3, no. 4, pp. 374–379, 2011. View at: Google Scholar
- J. Rochon, C. W. Bales, E. Ravussin et al., “Design and conduct of the CALERIE study: comprehensive assessment of the long-term effects of reducing intake of energy,” The Journals of Gerontology A, vol. 66, no. 1, pp. 97–108, 2011. View at: Publisher Site | Google Scholar
- C. K. Martin, S. K. Das, L. Lindblad et al., “Effect of calorie restriction on the free-living physical activity levels of nonobese humans: results of three randomized trials,” Journal of Applied Physiology, vol. 110, no. 4, pp. 956–963, 2011. View at: Publisher Site | Google Scholar
- K. Stein, A. Soare, T. E. Meyer, R. Cangemi, J. O. Holloszy, and L. Fontana, “Caloric restriction may reverse age-related autonomic decline in humans,” Aging Cell, vol. 11, no. 4, pp. 644–650, 2012. View at: Google Scholar
- R. M. Anderson and R. Weindruch, “Metabolic reprogramming, caloric restriction and aging,” Trends in Endocrinology and Metabolism, vol. 21, no. 3, pp. 134–141, 2010. View at: Publisher Site | Google Scholar
- J. M. Zahn, S. Poosala, A. B. Owen et al., “AGEMAP: a gene expression database for aging in mice,” PLoS Genetics, vol. 3, no. 11, p. e201, 2007. View at: Publisher Site | Google Scholar
- J. P. de Magalhães, J. Curado, and G. M. Church, “Meta-analysis of age-related gene expression profiles identifies common signatures of aging,” Bioinformatics, vol. 25, no. 7, pp. 875–881, 2009. View at: Publisher Site | Google Scholar
- S. K. Park and T. A. Prolla, “Lessons learned from gene expression profile studies of aging and caloric restriction,” Ageing Research Reviews, vol. 4, no. 1, pp. 55–65, 2005. View at: Publisher Site | Google Scholar
- G. S. Hotamisligil, “Inflammation and metabolic disorders,” Nature, vol. 444, no. 7121, pp. 860–867, 2006. View at: Publisher Site | Google Scholar
- F. Lago, C. Dieguez, J. Gómez-Reino, and O. Gualillo, “The emerging role of adipokines as mediators of inflammation and immune responses,” Cytokine & Growth Factor Reviews, vol. 18, no. 3-4, pp. 313–325, 2007. View at: Publisher Site | Google Scholar
- U. Meier and A. M. Gressner, “Endocrine regulation of energy metabolism: review of pathobiochemical and clinical chemical aspects of leptin, ghrelin, adiponectin, and resistin,” Clinical Chemistry, vol. 50, no. 9, pp. 1511–1525, 2004. View at: Publisher Site | Google Scholar
- M. Zhu, G. D. Lee, L. Ding et al., “Adipogenic signaling in rat white adipose tissue: modulation by aging and calorie restriction,” Experimental Gerontology, vol. 42, no. 8, pp. 733–744, 2007. View at: Publisher Site | Google Scholar
- K. Shinmura, K. Tamaki, K. Saito, Y. Nakano, T. Tobe, and R. Bolli, “Cardioprotective effects of short-term caloric restriction are mediated by adiponectin via activation of AMP-activated protein kinase,” Circulation, vol. 116, no. 24, pp. 2809–2817, 2007. View at: Publisher Site | Google Scholar
- J. J. Díez and P. Iglesias, “The role of the novel adipocyte-derived hormone adiponectin in human disease,” European Journal of Endocrinology, vol. 148, no. 3, pp. 293–300, 2003. View at: Publisher Site | Google Scholar
- O. Ukkola and M. Santaniemi, “Adiponectin: a link between excess adiposity and associated comorbidities?” Journal of Molecular Medicine, vol. 80, no. 11, pp. 696–702, 2002. View at: Publisher Site | Google Scholar
- O. Renaldi, B. Pramono, H. Sinorita, L. B. Purnomo, R. H. Asdie, and A. H. Asdie, “Hypoadiponectinemia: a risk factor for metabolic syndrome,” Acta medica Indonesiana, vol. 41, no. 1, pp. 20–24, 2009. View at: Google Scholar
- A. E. Civitarese, B. Ukropcova, S. Carling et al., “Role of adiponectin in human skeletal muscle bioenergetics,” Cell Metabolism, vol. 4, no. 1, pp. 75–87, 2006. View at: Publisher Site | Google Scholar
- M. Nishimura, Y. Izumiya, A. Higuchi et al., “Adiponectin prevents cerebral ischemic injury through endothelial nitric oxide synthase-dependent mechanisms,” Circulation, vol. 117, no. 2, pp. 216–223, 2008. View at: Publisher Site | Google Scholar
- J. M. Fernández-Real, A. López-Bermejo, R. Casamitjana, and W. Ricart, “Novel interactions of adiponectin with the endocrine system and inflammatory parameters,” The Journal of Clinical Endocrinology & Metabolism, vol. 88, no. 6, pp. 2714–2718, 2003. View at: Google Scholar
- W. Aldhahi and O. Hamdy, “Adipokines, inflammation, and the endothelium in diabetes,” Current Diabetes Reports, vol. 3, no. 4, pp. 293–298, 2003. View at: Google Scholar
- N. Ouchi, S. Kihara, T. Funahashi, Y. Matsuzawa, and K. Walsh, “Obesity, adiponectin and vascular inflammatory disease,” Current Opinion in Lipidology, vol. 14, no. 6, pp. 561–566, 2003. View at: Publisher Site | Google Scholar
- D. M. Huffman, D. R. Moellering, W. E. Grizzle, C. R. Stockard, M. S. Johnson, and T. R. Nagy, “Effect of exercise and calorie restriction on biomarkers of aging in mice,” American Journal of Physiology, vol. 294, no. 5, pp. R1618–R1627, 2008. View at: Publisher Site | Google Scholar
- L. Fontana, E. P. Weiss, D. T. Villareal, S. Klein, and J. O. Holloszy, “Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans,” Aging Cell, vol. 7, no. 5, pp. 681–687, 2008. View at: Publisher Site | Google Scholar
- A. R. Cameron, S. Anton, L. Melville et al., “Black tea polyphenols mimic insulin/insulin-like growth factor-1 signalling to the longevity factor FOXO1a,” Aging Cell, vol. 7, no. 1, pp. 69–77, 2008. View at: Publisher Site | Google Scholar
- W. Qin, W. Zhao, L. Ho et al., “Regulation of forkhead transcription factor FOXO3a contributes to calorie restriction-induced prevention of Alzheimer’s disease-type amyloid neuropathology and spatial memory deterioration,” Annals of the New York Academy of Sciences, vol. 1147, pp. 335–347, 2008. View at: Publisher Site | Google Scholar
- F. Flachsbart, A. Caliebe, R. Kleindorp et al., “Association of FOX03A variation with human longevity confirmed in German centenarians,” Proceedings of the National Academy of Sciences of the United States of America, vol. 106, no. 8, pp. 2700–2705, 2009. View at: Publisher Site | Google Scholar
- E. J. Anderson, M. E. Lustig, K. E. Boyle et al., “Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans,” The Journal of Clinical Investigation, vol. 119, no. 3, pp. 573–581, 2009. View at: Publisher Site | Google Scholar
- D. E. Harrison, R. Strong, Z. D. Sharp et al., “Rapamycin fed late in life extends lifespan in genetically heterogeneous mice,” Nature, vol. 460, no. 7253, pp. 392–395, 2009. View at: Publisher Site | Google Scholar
- C. Chen, Y. Liu, Y. Liu, and P. Zheng, “mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells,” Science Signaling, vol. 2, no. 98, p. ra75, 2009. View at: Publisher Site | Google Scholar
- P. Kapahi, D. Chen, A. N. Rogers et al., “With TOR, less is more: a key role for the conserved nutrient-sensing TOR pathway in aging,” Cell Metabolism, vol. 11, no. 6, pp. 453–465, 2010. View at: Publisher Site | Google Scholar
- B. M. Zid, A. N. Rogers, S. D. Katewa et al., “4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila,” Cell, vol. 139, no. 1, pp. 149–160, 2009. View at: Publisher Site | Google Scholar
- S. Sengupta, T. R. Peterson, M. Laplante, S. Oh, and D. M. Sabatini, “mTORC1 controls fasting-induced ketogenesis and its modulation by ageing,” Nature, vol. 468, no. 7327, pp. 1100–1106, 2010. View at: Publisher Site | Google Scholar
- J. T. Cunningham, J. T. Rodgers, D. H. Arlow, F. Vazquez, V. K. Mootha, and P. Puigserver, “mTOR controls mitochondrial oxidative function through a YY1-PGC-1α transcriptional complex,” Nature, vol. 450, no. 7170, pp. 736–740, 2007. View at: Publisher Site | Google Scholar
- A. Ramanathan and S. L. Schreiber, “Direct control of mitochondrial function by mTOR,” Proceedings of the National Academy of Sciences of the United States of America, vol. 106, no. 52, pp. 22229–22232, 2009. View at: Publisher Site | Google Scholar
- J. Knapowski, K. Wieczorowska-Tobis, and J. Witowski, “Pathophysiology of ageing,” Journal of Physiology and Pharmacology, vol. 53, no. 2, pp. 135–146, 2002. View at: Google Scholar
- J. P. J. Issa, N. Ahuja, M. Toyota, M. P. Bronner, and T. A. Brentnall, “Accelerated age-related CpG island methylation in ulcerative colitis,” Cancer Research, vol. 61, no. 9, pp. 3573–3577, 2001. View at: Google Scholar
- J. P. J. Issa, Y. L. Ottaviano, P. Celano, S. R. Hamilton, N. E. Davidson, and S. B. Baylin, “Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon,” Nature Genetics, vol. 7, no. 4, pp. 536–540, 1994. View at: Publisher Site | Google Scholar
- J. P. J. Issa, P. M. Vertino, C. D. Boehm, I. F. Newsham, and S. B. Baylin, “Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis,” Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no. 21, pp. 11757–11762, 1996. View at: Publisher Site | Google Scholar
- R. P. Singhal, L. L. Mays-Hoopes, and G. L. Eichhorn, “DNA methylation in aging of mice,” Mechanisms of Ageing and Development, vol. 41, no. 3, pp. 199–210, 1987. View at: Google Scholar
- T. Waki, G. Tamura, M. Sato, and T. Motoyama, “Age-related methylation of tumor suppressor and tumor-related genes: an analysis of autopsy samples,” Oncogene, vol. 22, no. 26, pp. 4128–4133, 2003. View at: Publisher Site | Google Scholar
- V. L. Wilson, R. A. Smith, S. Ma, and R. G. Cutler, “Genomic 5-methyldeoxycytidine decreases with age,” The Journal of Biological Chemistry, vol. 262, no. 21, pp. 9948–9951, 1987. View at: Google Scholar
- T. Y. Kim, H. J. Lee, K. S. Hwang et al., “Methylation of RUNX3 in various types of human cancers and premalignant stages of gastric carcinoma,” Laboratory Investigation, vol. 84, no. 4, pp. 479–484, 2004. View at: Publisher Site | Google Scholar
- Y. Li, M. Daniel, and T. O. Tollefsbol, “Epigenetic regulation of caloric restriction in aging,” BMC Medicine, vol. 9, article 98, 2011. View at: Google Scholar
- A. Vaquero and D. Reinberg, “Calorie restriction and the exercise of chromatin,” Genes & Development, vol. 23, no. 16, pp. 1849–1869, 2009. View at: Publisher Site | Google Scholar
- U. Muñoz-Najar and J. M. Sedivy, “Epigenetic control of aging,” Antioxidants & Redox Signaling, vol. 14, no. 2, pp. 241–259, 2011. View at: Publisher Site | Google Scholar
- B. S. Hass, R. W. Hart, M. H. Lu, and B. D. Lyn-Cook, “Effects of caloric restriction in animals on cellular function, oncogene expression, and DNA methylation in vitro,” Mutation Research, vol. 295, no. 4–6, pp. 281–289, 1993. View at: Google Scholar
- Y. Li, L. Liu, and T. O. Tollefsbol, “Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression,” The FASEB Journal, vol. 24, no. 5, pp. 1442–1453, 2010. View at: Publisher Site | Google Scholar
- R. S. Ahima, “Connecting obesity, aging and diabetes,” Nature Medicine, vol. 15, no. 9, pp. 996–997, 2009. View at: Publisher Site | Google Scholar
- T. M. Larsen, S. Dalskov, M. Van Baak et al., “The diet, obesity and genes (diogenes) dietary study in eight European countries—a comprehensive design for long-term intervention,” Obesity Reviews, vol. 11, no. 1, pp. 76–91, 2010. View at: Publisher Site | Google Scholar
- F. I. Milagro, J. Campión, P. Cordero et al., “A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss,” The FASEB Journal, vol. 25, no. 4, pp. 1378–1389, 2011. View at: Publisher Site | Google Scholar
- L. Bouchard, R. Rabasa-Lhoret, M. Faraj et al., “Differential epigenomic and transcriptomic responses in subcutaneous adipose tissue between low and high responders to caloric restriction,” The American Journal of Clinical Nutrition, vol. 91, no. 2, pp. 309–320, 2010. View at: Publisher Site | Google Scholar
- J. Campión, F. I. Milagro, E. Goyenechea, and J. A. Martínez, “TNF-α promoter methylation as a predictive biomarker for weight-loss response,” Obesity, vol. 17, no. 6, pp. 1293–1297, 2009. View at: Publisher Site | Google Scholar
- A. L. Clayton, C. A. Hazzalin, and L. C. Mahadevan, “Enhanced histone acetylation and transcription: a dynamic perspective,” Molecular Cell, vol. 23, no. 3, pp. 289–296, 2006. View at: Publisher Site | Google Scholar
- M. Meyerson, C. M. Counter, E. N. Eaton et al., “hEST2, the putative human telomerase catalytic subunit gene, is up- regulated in tumor cells and during immortalization,” Cell, vol. 90, no. 4, pp. 785–795, 1997. View at: Publisher Site | Google Scholar
- T. Kanaya, S. Kyo, M. Takakura, H. Ito, M. Namiki, and M. Inoue, “hTERT is a critical determinant of telomerase activity in renal-cell carcinoma,” International Journal of Cancer, vol. 78, no. 5, pp. 539–543, 1998. View at: Google Scholar
- S. J. Lin, P. A. Defossez, and L. Guarente, “Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae,” Science, vol. 289, no. 5487, pp. 2126–2128, 2000. View at: Publisher Site | Google Scholar
- L. Guarente and F. Picard, “Calorie restriction—the SIR2 connection,” Cell, vol. 120, no. 4, pp. 473–482, 2005. View at: Publisher Site | Google Scholar
- I. B. Leibiger and P. O. Berggren, “Sirt1: a metabolic master switch that modulates lifespan,” Nature Medicine, vol. 12, no. 1, pp. 34–36, 2006. View at: Publisher Site | Google Scholar
- L. Bordone, D. Cohen, A. Robinson et al., “SIRT1 transgenic mice show phenotypes resembling calorie restriction,” Aging Cell, vol. 6, no. 6, pp. 759–767, 2007. View at: Publisher Site | Google Scholar
- H. Y. Cohen, C. Miller, K. J. Bitterman et al., “Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase,” Science, vol. 305, no. 5682, pp. 390–392, 2004. View at: Publisher Site | Google Scholar
- Y. Kanfi, V. Peshti, Y. M. Gozlan, M. Rathaus, R. Gil, and H. Y. Cohen, “Regulation of SIRT1 protein levels by nutrient availability,” FEBS Letters, vol. 582, no. 16, pp. 2417–2423, 2008. View at: Publisher Site | Google Scholar
- A. B. Crujeiras, D. Parra, E. Goyenechea, and J. A. Martínez, “Sirtuin gene expression in human mononuclear cells is modulated by caloric restriction,” European Journal of Clinical Investigation, vol. 38, no. 9, pp. 672–678, 2008. View at: Publisher Site | Google Scholar
- L. A. Wakeling, L. J. Ions, and D. Ford, “Could Sirt1-mediated epigenetic effects contribute to the longevity response to dietary restriction and be mimicked by other dietary interventions?” Age, vol. 31, no. 4, pp. 327–341, 2009. View at: Publisher Site | Google Scholar
- Y. Li and T. O. Tollefsbol, “P16INK4a suppression by glucose restriction contributes to human cellular lifespan extension through SIRT1-mediated epigenetic and genetic mechanisms,” PLoS ONE, vol. 6, no. 2, Article ID e17421, 2011. View at: Publisher Site | Google Scholar
- M. C. Haigis and L. P. Guarente, “Mammalian sirtuins—emerging roles in physiology, aging, and calorie restriction,” Genes & Development, vol. 20, no. 21, pp. 2913–2921, 2006. View at: Publisher Site | Google Scholar
- J. Luo, A. Y. Nikolaev, S. I. Imai et al., “Negative control of p53 by Sir2α promotes cell survival under stress,” Cell, vol. 107, no. 2, pp. 137–148, 2001. View at: Publisher Site | Google Scholar
- E. Langley, M. Pearson, M. Faretta et al., “Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence,” The EMBO Journal, vol. 21, no. 10, pp. 2383–2396, 2002. View at: Publisher Site | Google Scholar
- H. Vaziri, S. K. Dessain, E. N. Eaton et al., “hSIR2SIRT1 functions as an NAD-dependent p53 deacetylase,” Cell, vol. 107, no. 2, pp. 149–159, 2001. View at: Publisher Site | Google Scholar
- A. Brunet, L. B. Sweeney, J. F. Sturgill et al., “Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase,” Science, vol. 303, no. 5666, pp. 2011–2015, 2004. View at: Publisher Site | Google Scholar
- M. C. Motta, N. Divecha, M. Lemieux et al., “Mammalian SIRT1 represses forkhead transcription factors,” Cell, vol. 116, no. 4, pp. 551–563, 2004. View at: Publisher Site | Google Scholar
- M. M. Schilling, J. K. Oeser, J. N. Boustead, B. P. Flemming, and R. M. O’Brien, “Gluconeogenesis: re-evaluating the FOXO1-PGC-1α connection,” Nature, vol. 443, no. 7111, pp. E10–E11, 2006. View at: Publisher Site | Google Scholar
- R. B. Vega, J. M. Huss, and D. P. Kelly, “The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor α in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes,” Molecular and Cellular Biology, vol. 20, no. 5, pp. 1868–1876, 2000. View at: Publisher Site | Google Scholar
- J. Koubova and L. Guarente, “How does calorie restriction work?” Genes & Development, vol. 17, no. 3, pp. 313–321, 2003. View at: Publisher Site | Google Scholar
- R. S. Sohal and R. Weindruch, “Oxidative stress, caloric restriction, and aging,” Science, vol. 273, no. 5271, pp. 59–63, 1996. View at: Google Scholar
- B. J. Merry, “Molecular mechanisms linking calorie restriction and longevity,” The International Journal of Biochemistry & Cell Biology, vol. 34, no. 11, pp. 1340–1354, 2002. View at: Publisher Site | Google Scholar
- J. Jeong, K. Juhn, H. Lee et al., “SIRT1 promotes DNA repair activity and deacetylation of Ku70,” Experimental & Molecular Medicine, vol. 39, no. 1, pp. 8–13, 2007. View at: Google Scholar
- H. Y. Cohen, S. Lavu, K. J. Bitterman et al., “Acetylation of the C terminus of Ku70 by CBP and PCAF controls Bax-mediated apoptosis,” Molecular Cell, vol. 13, no. 5, pp. 627–638, 2004. View at: Publisher Site | Google Scholar
- H. Wong and K. Riabowol, “Differential CDK-inhibitor gene expression in aging human diploid fibroblasts,” Experimental Gerontology, vol. 31, no. 1-2, pp. 311–325, 1996. View at: Publisher Site | Google Scholar
- J. Gil and G. Peters, “Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all,” Nature Reviews, vol. 7, no. 9, pp. 667–677, 2006. View at: Publisher Site | Google Scholar
- J. Krishnamurthy, C. Torrice, M. R. Ramsey et al., “Ink4a/Arf expression is a biomarker of aging,” The Journal of Clinical Investigation, vol. 114, no. 9, pp. 1299–1307, 2004. View at: Publisher Site | Google Scholar
- D. A. Alcorta, Y. Xiong, D. Phelps, G. Hannon, D. Beach, and J. C. Barrett, “Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts,” Proceedings of the National Academy of Sciences of the United States of America, vol. 93, no. 24, pp. 13742–13747, 1996. View at: Publisher Site | Google Scholar
- A. Melk, B. M. W. Schmidt, O. Takeuchi, B. Sawitzki, D. C. Rayner, and P. F. Halloran, “Expression of p16INK4a and other cell cycle regulator and senescence associated genes in aging human kidney,” Kidney International, vol. 65, no. 2, pp. 510–520, 2004. View at: Publisher Site | Google Scholar
- W. Fischle, Y. Wang, and C. D. Allis, “Histone and chromatin cross-talk,” Current Opinion in Cell Biology, vol. 15, no. 2, pp. 172–183, 2003. View at: Publisher Site | Google Scholar
- T. Kouzarides, “Histone methylation in transcriptional control,” Current Opinion in Genetics & Development, vol. 12, no. 2, pp. 198–209, 2002. View at: Publisher Site | Google Scholar
- N. Noren Hooten, K. Abdelmohsen, M. Gorospe, N. Ejiogu, A. B. Zonderman, and M. K. Evans, “microRNA expression patterns reveal differential expression of target genes with age,” PloS One, vol. 5, no. 5, Article ID e10724, 2010. View at: Publisher Site | Google Scholar
- A. De Lencastre, Z. Pincus, K. Zhou, M. Kato, S. S. Lee, and F. J. Slack, “MicroRNAs both promote and antagonize longevity in C. elegans,” Current Biology, vol. 20, no. 24, pp. 2159–2168, 2010. View at: Publisher Site | Google Scholar
- D. J. Bates, N. Li, R. Liang et al., “MicroRNA regulation in Ames dwarf mouse liver may contribute to delayed aging,” Aging Cell, vol. 9, no. 1, pp. 1–18, 2010. View at: Publisher Site | Google Scholar
- U. A. Ørom, M. K. Lim, J. E. Savage et al., “MicroRNA-203 regulates caveolin-1 in breast tissue during caloric restriction,” Cell Cycle, vol. 11, no. 7, pp. 1291–1295, 2012. View at: Google Scholar
- A. Khanna, S. Muthusamy, R. Liang, H. Sarojini, and E. Wang, “Gain of survival signaling by down-regulation of three key miRNAs in brain of calorie-restricted mice,” Aging, vol. 3, no. 3, pp. 223–236, 2011. View at: Google Scholar
- E. Paradis, H. Douillard, M. Koutroumanis, C. Goodyer, and A. LeBlanc, “Amyloid β peptide of Alzheimer’s disease downregulates bcl-2 and upregulates bax expression in human neurons,” Journal of Neuroscience, vol. 16, no. 23, pp. 7533–7539, 1996. View at: Google Scholar
- C. Perier, J. Bové, D. C. Wu et al., “Two molecular pathways initiate mitochondria-dependent dopaminergic neurodegeneration in experimental Parkinson’s disease,” Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no. 19, pp. 8161–8166, 2007. View at: Publisher Site | Google Scholar
- N. Louneva, J. W. Cohen, L. Y. Han et al., “Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease,” The American Journal of Pathology, vol. 173, no. 5, pp. 1488–1495, 2008. View at: Publisher Site | Google Scholar
- M. Yamada, K. Kida, W. Amutuhaire, F. Ichinose, and M. Kaneki, “Gene disruption of caspase-3 prevents MPTP-induced Parkinson’s disease in mice,” Biochemical and Biophysical Research Communications, vol. 402, no. 2, pp. 312–318, 2010. View at: Publisher Site | Google Scholar
- W. Kudo, H. P. Lee, M. A. Smith, X. Zhu, S. Matsuyama, and H. G. Lee, “Inhibition of Bax protects neuronal cells from oligomeric Aβ neurotoxicity,” Cell Death & Disease, vol. 3, Article ID e309, 2012. View at: Publisher Site | Google Scholar
- D. K. Ingram, M. Zhu, J. Mamczarz et al., “Calorie restriction mimetics: an emerging research field,” Aging Cell, vol. 5, no. 2, pp. 97–108, 2006. View at: Publisher Site | Google Scholar
- H. R. Warner, D. Ingram, R. A. Miller, N. L. Nadon, and A. G. Richardson, “Program for testing biological interventions to promote healthy aging,” Mechanisms of Ageing and Development, vol. 115, no. 3, pp. 199–207, 2000. View at: Publisher Site | Google Scholar
- N. L. Nadon, R. Strong, R. A. Miller et al., “Design of aging intervention studies: the NIA interventions testing program,” Age, vol. 30, no. 4, pp. 187–199, 2008. View at: Publisher Site | Google Scholar
- R. A. Miller, D. E. Harrison, C. M. Astle et al., “An aging interventions testing program: study design and interim report,” Aging Cell, vol. 6, no. 4, pp. 565–575, 2007. View at: Publisher Site | Google Scholar
- R. Strong, R. A. Miller, C. M. Astle et al., “Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice,” Aging Cell, vol. 7, no. 5, pp. 641–650, 2008. View at: Publisher Site | Google Scholar
- N. L. Nadon, “Exploiting the rodent model for studies on the pharmacology of lifespan extension,” Aging Cell, vol. 5, no. 1, pp. 9–15, 2006. View at: Publisher Site | Google Scholar
- J. O. Holloszy, “Mortality rate and longevity of food-restricted exercising male rats: a reevaluation,” Journal of Applied Physiology, vol. 82, no. 2, pp. 399–403, 1997. View at: Google Scholar
- K. C. Deruisseau, A. N. Kavazis, S. Judge et al., “Moderate caloric restriction increases diaphragmatic antioxidant enzyme mRNA, but not when combined with lifelong exercise,” Antioxidants & Redox Signaling, vol. 8, no. 3-4, pp. 539–547, 2006. View at: Publisher Site | Google Scholar
- A. Y. Seo, T. Hofer, B. Sung, S. Judge, H. Y. Chung, and C. Leeuwenburgh, “Hepatic oxidative stress during aging: effects of 8% long-term calorie restriction and lifelong exercise,” Antioxidants & Redox Signaling, vol. 8, no. 3-4, pp. 529–538, 2006. View at: Publisher Site | Google Scholar
- R. Kalani, S. Judge, C. Carter, M. Pahor, and C. Leeuwenburgh, “Effects of caloric restriction and exercise on age-related, chronic inflammation assessed by C-reactive protein and interleukin-6,” The Journals of Gerontology A, vol. 61, no. 3, pp. 211–217, 2006. View at: Google Scholar
- P. Abete, G. Testa, G. Galizia et al., “Tandem action of exercise training and food restriction completely preserves ischemic preconditioning in the aging heart,” Experimental Gerontology, vol. 40, no. 1-2, pp. 43–50, 2005. View at: Publisher Site | Google Scholar
- D. L. Crandall, R. P. Feirer, D. R. Griffith, and D. C. Beitz, “Relative role of caloric restriction and exercise training upon susceptibility to isoproterenol-induced myocardial infarction in male rats,” The American Journal of Clinical Nutrition, vol. 34, no. 5, pp. 841–847, 1981. View at: Google Scholar
- A. E. Civitarese, S. Carling, L. K. Heilbronn et al., “Calorie restriction increases muscle mitochondrial biogenesis in healthy humans,” PLoS Medicine, vol. 4, no. 3, article e76, 2007. View at: Publisher Site | Google Scholar
- D. E. Larson-Meyer, B. R. Newcomer, L. K. Heilbronn et al., “Effect of 6-month calorie restriction and exercise on serum and liver lipids and markers of liver function,” Obesity, vol. 16, no. 6, pp. 1355–1362, 2008. View at: Publisher Site | Google Scholar
- M. Lefevre, L. M. Redman, L. K. Heilbronn et al., “Caloric restriction alone and with exercise improves CVD risk in healthy non-obese individuals,” Atherosclerosis, vol. 203, no. 1, pp. 206–213, 2009. View at: Publisher Site | Google Scholar
- D. E. Larson-Meyer, L. Redman, L. K. Heilbronn, C. K. Martin, and E. Ravussin, “Caloric restriction with or without exercise: the fitness versus fatness debate,” Medicine and Science in Sports and Exercise, vol. 42, no. 1, pp. 152–159, 2010. View at: Publisher Site | Google Scholar
- N. E. Silverman, B. J. Nicklas, and A. S. Ryan, “Addition of aerobic exercise to a weight loss program increases BMD, with an associated reduction in inflammation in overweight postmenopausal women,” Calcified Tissue International, vol. 84, no. 4, pp. 257–265, 2009. View at: Publisher Site | Google Scholar
- J. F. Trepanowski, R. E. Canale, K. E. Marshall, M. M. Kabir, and R. J. Bloomer, “Impact of caloric and dietary restriction regimens on markers of health and longevity in humans and animals: a summary of available findings,” Nutrition Journal, vol. 10, article 107, 2011. View at: Google Scholar
- K. Iwasaki, C. A. Gleiser, E. J. Masoro, C. A. McMahan, E. Seo, and B. P. Yu, “The influence of dietary protein source on longevity and age-related disease processes of Fischer rats,” Journals of Gerontology, vol. 43, no. 1, pp. B5–B12, 1988. View at: Google Scholar
- I. Shimokawa, Y. Higami, B. P. Yu, E. J. Masoro, and T. Ikeda, “Influence of dietary components on occurrence of and mortality due to neoplasms in male F344 rats,” Aging, vol. 8, no. 4, pp. 254–262, 1996. View at: Google Scholar
- M. Khorakova, Z. Deil, D. Khausman, and K. Matsek, “Effect of carbohydrate-enriched diet and subsequent food restriction on life prolongation in Fischer 344 male rats,” Fiziologicheskii Zhurnal, vol. 36, no. 5, pp. 16–21, 1990. View at: Google Scholar
- C. Kubo, B. C. Johnson, A. Gajjar, and R. A. Good, “Crucial dietary factors in maximizing life span and longevity in autoimmune-prone mice,” The Journal of Nutrition, vol. 117, no. 6, pp. 1129–1135, 1987. View at: Google Scholar
- R. Pamplona and G. Barja, “Mitochondrial oxidative stress, aging and caloric restriction: the protein and methionine connection,” Biochimica et Biophysica Acta, vol. 1757, no. 5-6, pp. 496–508, 2006. View at: Publisher Site | Google Scholar
- A. Sanz, P. Caro, J. G. Sanchez, and G. Barja, “Effect of lipid restriction on mitochondrial free radical production and oxidative DNA damage,” Annals of the New York Academy of Sciences, vol. 1067, no. 1, pp. 200–209, 2006. View at: Publisher Site | Google Scholar
- A. Sanz, J. Gómez, P. Caro, and G. Barja, “Carbohydrate restriction does not change mitochondrial free radical generation and oxidative DNA damage,” Journal of Bioenergetics and Biomembranes, vol. 38, no. 5-6, pp. 327–333, 2006. View at: Publisher Site | Google Scholar
- P. E. Segall and P. S. Timiras, “Pathophysiologic findings after chronic tryptophan deficiency in rats: a model for delayed growth and aging,” Mechanisms of Ageing and Development, vol. 5, no. 2, pp. 109–124, 1976. View at: Google Scholar
- H. Ooka, P. E. Segall, and P. S. Timiras, “Histology and survival in age-delayed low-tryptophan-fed rats,” Mechanisms of Ageing and Development, vol. 43, no. 1, pp. 79–98, 1988. View at: Google Scholar
- R. A. Miller, G. Buehner, Y. Chang, J. M. Harper, R. Sigler, and M. Smith-Wheelock, “Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance,” Aging Cell, vol. 4, no. 3, pp. 119–125, 2005. View at: Publisher Site | Google Scholar
- N. Orentreich, J. R. Matias, A. DeFelice, and J. A. Zimmerman, “Low methionine ingestion by rats extends life span,” The Journal of Nutrition, vol. 123, no. 2, pp. 269–274, 1993. View at: Google Scholar
- J. P. Richie Jr., Y. Leutzinger, S. Parthasarathy, V. Malloy, N. Orentreich, and J. A. Zimmerman, “Methionine restriction increases blood glutathione and longevity in F344 rats,” The FASEB Journal, vol. 8, no. 15, pp. 1302–1307, 1994. View at: Google Scholar
- J. P. Richie Jr., D. Komninou, Y. Leutzinger et al., “Tissue glutathione and cysteine levels in methionine-restricted rats,” Nutrition, vol. 20, no. 9, pp. 800–805, 2004. View at: Publisher Site | Google Scholar
- J. A. Zimmerman, V. Malloy, R. Krajcik, and N. Orentreich, “Nutritional control of aging,” Experimental Gerontology, vol. 38, no. 1-2, pp. 47–52, 2003. View at: Publisher Site | Google Scholar
- A. Sanz, P. Caro, V. Ayala, M. Portero-Otin, R. Pamplona, and G. Barja, “Methionine restriction decreases mitochondrial oxygen radical generation and leak as well as oxidative damage to mitochondrial DNA and proteins,” The FASEB Journal, vol. 20, no. 8, pp. 1064–1073, 2006. View at: Publisher Site | Google Scholar
- P. Caro, J. Gómez, M. López-Torres et al., “Forty percent and eighty percent methionine restriction decrease mitochondrial ROS generation and oxidative stress in rat liver,” Biogerontology, vol. 9, no. 3, pp. 183–196, 2008. View at: Publisher Site | Google Scholar
- M. C. Ruiz, V. Ayala, M. Portero-Otín, J. R. Requena, G. Barja, and R. Pamplona, “Protein methionine content and MDA-lysine adducts are inversely related to maximum life span in the heart of mammals,” Mechanisms of Ageing and Development, vol. 126, no. 10, pp. 1106–1114, 2005. View at: Publisher Site | Google Scholar
- N. Hidiroglou, G. S. Gilani, L. Long et al., “The influence of dietary vitamin E, fat, and methionine on blood cholesterol profile, homocysteine levels, and oxidizability of low density lipoprotein in the gerbil,” The Journal of Nutritional Biochemistry, vol. 15, no. 12, pp. 730–740, 2004. View at: Publisher Site | Google Scholar
- P. Caro, J. Gomez, I. Sanchez et al., “Effect of 40% restriction of dietary amino acids (except methionine) on mitochondrial oxidative stress and biogenesis, AIF and SIRT1 in rat liver,” Biogerontology, vol. 10, no. 5, pp. 579–592, 2009. View at: Publisher Site | Google Scholar
- M. F. McCarty, J. Barroso-Aranda, and F. Contreras, “The low-methionine content of vegan diets may make methionine restriction feasible as a life extension strategy,” Medical Hypotheses, vol. 72, no. 2, pp. 125–128, 2009. View at: Publisher Site | Google Scholar
- K. A. Varady and M. K. Hellerstein, “Alternate-day fasting and chronic disease prevention: a review of human and animal trials,” The American Journal of Clinical Nutrition, vol. 86, no. 1, pp. 7–13, 2007. View at: Google Scholar
- R. M. Anson, Z. Guo, R. de Cabo et al., “Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake,” Proceedings of the National Academy of Sciences of the United States of America, vol. 100, no. 10, pp. 6216–6220, 2003. View at: Publisher Site | Google Scholar
- O. Descamps, J. Riondel, V. Ducros, and A. M. Roussel, “Mitochondrial production of reactive oxygen species and incidence of age-associated lymphoma in OF1 mice: effect of alternate-day fasting,” Mechanisms of Ageing and Development, vol. 126, no. 11, pp. 1185–1191, 2005. View at: Publisher Site | Google Scholar
- W. Duan, Z. Guo, H. Jiang, M. Ware, and M. P. Mattson, “Reversal of behavioral and metabolic abnormalities, and insulin resistance syndrome, by dietary restriction in mice deficient in brain-derived neurotrophic factor,” Endocrinology, vol. 144, no. 6, pp. 2446–2453, 2003. View at: Publisher Site | Google Scholar
- I. Ahmet, R. Wan, M. P. Mattson, E. G. Lakatta, and M. Talan, “Cardioprotection by intermittent fasting in rats,” Circulation, vol. 112, no. 20, pp. 3115–3121, 2005. View at: Publisher Site | Google Scholar
- D. E. Mager, R. Wan, M. Brown et al., “Caloric restriction and intermittent fasting alter spectral measures of heart rate and blood pressure variability in rats,” The FASEB Journal, vol. 20, no. 6, pp. 631–637, 2006. View at: Publisher Site | Google Scholar
- C. R. Pedersen, I. Hagemann, T. Bock, and K. Buschard, “Intermittent feeding and fasting reduces diabetes incidence in BB rats,” Autoimmunity, vol. 30, no. 4, pp. 243–250, 1999. View at: Google Scholar
- K. Tikoo, D. N. Tripathi, D. G. Kabra, V. Sharma, and A. B. Gaikwad, “Intermittent fasting prevents the progression of type I diabetic nephropathy in rats and changes the expression of Sir2 and p53,” FEBS Letters, vol. 581, no. 5, pp. 1071–1078, 2007. View at: Publisher Site | Google Scholar
- R. Wan, S. Camandola, and M. P. Mattson, “Intermittent fasting and dietary supplementation with 2-deoxy-D-glucose improve functional and metabolic cardiovascular risk factors in rats,” The FASEB Journal, vol. 17, no. 9, pp. 1133–1134, 2003. View at: Google Scholar
- L. K. Heilbronn, S. R. Smith, C. K. Martin, S. D. Anton, and E. Ravussin, “Alternate-day fasting in nonobese subjects: effects on body weight, body composition, and energy metabolism,” The American Journal of Clinical Nutrition, vol. 81, no. 1, pp. 69–73, 2005. View at: Google Scholar
- N. Halberg, M. Henriksen, N. Söderhamn et al., “Effect of intermittent fasting and refeeding on insulin action in healthy men,” Journal of Applied Physiology, vol. 99, no. 6, pp. 2128–2136, 2005. View at: Publisher Site | Google Scholar
- J. B. Johnson, W. Summer, R. G. Cutler et al., “Alternate day calorie restriction improves clinical findings and reduces markers of oxidative stress and inflammation in overweight adults with moderate asthma,” Free Radical Biology and Medicine, vol. 42, no. 5, pp. 665–674, 2007. View at: Publisher Site | Google Scholar
- V. D. Dixit, H. Yang, K. S. Sayeed et al., “Controlled meal frequency without caloric restriction alters peripheral blood mononuclear cell cytokine production,” Journal of Inflammation, vol. 8, article 6, 2011. View at: Publisher Site | Google Scholar
- K. T. Howitz, K. J. Bitterman, H. Y. Cohen et al., “Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan,” Nature, vol. 425, no. 6954, pp. 191–196, 2003. View at: Publisher Site | Google Scholar
- J. G. Wood, B. Rogina, S. Lavu et al., “Sirtuin activators mimic caloric restriction and delay ageing in metazoans,” Nature, vol. 430, no. 7000, pp. 686–689, 2004. View at: Google Scholar
- H. Yang, J. A. Baur, A. Chen, C. Miller, and D. A. Sinclair, “Design and synthesis of compounds that extend yeast replicative lifespan,” Aging Cell, vol. 6, no. 1, pp. 35–43, 2007. View at: Publisher Site | Google Scholar
- D. R. Valenzano, E. Terzibasi, T. Genade, A. Cattaneo, L. Domenici, and A. Cellerino, “Resveratrol prolongs lifespan and retards the onset of age-related markers in a short-lived vertebrate,” Current Biology, vol. 16, no. 3, pp. 296–300, 2006. View at: Publisher Site | Google Scholar
- M. Kaeberlein, T. McDonagh, B. Heltweg et al., “Substrate-specific activation of sirtuins by resveratrol,” The Journal of Biological Chemistry, vol. 280, no. 17, pp. 17038–17045, 2005. View at: Publisher Site | Google Scholar
- M. Kaeberlein and R. W. Powers III, “Sir2 and calorie restriction in yeast: a skeptical perspective,” Ageing Research Reviews, vol. 6, no. 2, pp. 128–140, 2007. View at: Publisher Site | Google Scholar
- M. Kaeberlein and B. K. Kennedy, “Does resveratrol activate yeast Sir2 in vivo?” Aging Cell, vol. 6, no. 4, pp. 415–416, 2007. View at: Publisher Site | Google Scholar
- D. L. Smith Jr., C. Li, M. Matecic, N. Maqani, M. Bryk, and J. S. Smith, “Calorie restriction effects on silencing and recombination at the yeast rDNA,” Aging Cell, vol. 8, no. 6, pp. 633–642, 2009. View at: Publisher Site | Google Scholar
- T. M. Bass, D. Weinkove, K. Houthoofd, D. Gems, and L. Partridge, “Effects of resveratrol on lifespan in Drosophila melanogaster and Caenorhabditis elegans,” Mechanisms of Ageing and Development, vol. 128, no. 10, pp. 546–552, 2007. View at: Publisher Site | Google Scholar
- E. L. Greer and A. Brunet, “Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans,” Aging Cell, vol. 8, no. 2, pp. 113–127, 2009. View at: Publisher Site | Google Scholar
- T. L. Kaeberlein, E. D. Smith, M. Tsuchiya et al., “Lifespan extension in Caenorhabditis elegans by complete removal of food,” Aging Cell, vol. 5, no. 6, pp. 487–494, 2006. View at: Publisher Site | Google Scholar
- S. Zou, J. R. Carey, P. Liedo et al., “The prolongevity effect of resveratrol depends on dietary composition and calorie intake in a tephritid fruit fly,” Experimental Gerontology, vol. 44, no. 6-7, pp. 472–476, 2009. View at: Publisher Site | Google Scholar
- M. Riesen and A. Morgan, “Calorie restriction reduces rDNA recombination independently of rDNA silencing,” Aging Cell, vol. 8, no. 6, pp. 624–632, 2009. View at: Publisher Site | Google Scholar
- M. Pacholec, J. E. Bleasdale, B. Chrunyk et al., “SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1,” The Journal of Biological Chemistry, vol. 285, no. 11, pp. 8340–8351, 2010. View at: Publisher Site | Google Scholar
- J. A. Baur, K. J. Pearson, N. L. Price et al., “Resveratrol improves health and survival of mice on a high-calorie diet,” Nature, vol. 444, no. 7117, pp. 337–342, 2006. View at: Publisher Site | Google Scholar
- M. Zang, S. Xu, K. A. Maitland-Toolan et al., “Polyphenols stimulate AMP-activated protein kinase, lower lipids, and inhibit accelerated atherosclerosis in diabetic LDL receptor-deficient mice,” Diabetes, vol. 55, no. 8, pp. 2180–2191, 2006. View at: Publisher Site | Google Scholar
- B. Dasgupta and J. Milbrandt, “Resveratrol stimulates AMP kinase activity in neurons,” Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no. 17, pp. 7217–7222, 2007. View at: Publisher Site | Google Scholar
- N. L. Price, A. P. Gomes, A. J. Ling et al., “SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function,” Cell Metabolism, vol. 15, no. 5, pp. 675–690, 2012. View at: Google Scholar
- J. L. Barger, T. Kayo, J. M. Vann et al., “A low dose of dietary resveratrol partially mimics caloric restriction and retards aging parameters in mice,” PLoS ONE, vol. 3, no. 6, Article ID e2264, 2008. View at: Publisher Site | Google Scholar
- K. J. Pearson, J. A. Baur, K. N. Lewis et al., “Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span,” Cell Metabolism, vol. 8, no. 2, pp. 157–168, 2008. View at: Publisher Site | Google Scholar
- R. A. Miller, D. E. Harrison, C. M. Astle et al., “Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice,” The Journals of Gerontology A, vol. 66, no. 2, pp. 191–201, 2011. View at: Publisher Site | Google Scholar
- D. L. Smith Jr., T. R. Nagy, and D. B. Allison, “Calorie restriction: what recent results suggest for the future of ageing research,” European Journal of Clinical Investigation, vol. 40, no. 5, pp. 440–450, 2010. View at: Publisher Site | Google Scholar
- A. Dal-Pan, S. Blanc, and F. Aujard, “Resveratrol suppresses body mass gain in a seasonal non-human primate model of obesity,” BMC Physiology, vol. 10, no. 1, article 11, 2010. View at: Publisher Site | Google Scholar
- A. Dal-Pan, J. Terrien, F. Pifferi et al., “Caloric restriction or resveratrol supplementation and ageing in a non-human primate: first-year outcome of the RESTRIKAL study in Microcebus murinus,” Age, vol. 33, no. 1, pp. 15–31, 2011. View at: Publisher Site | Google Scholar
- L. Peltz, J. Gomez, M. Marquez et al., “Resveratrol exerts dosage and duration dependent effect on human mesenchymal stem cell development,” PLoS One, vol. 7, no. 5, Article ID e37162, 2012. View at: Google Scholar
- J. Mikuła-Pietrasik, A. Kuczmarska, B. Rubiś et al., “Resveratrol delays replicative senescence of human mesothelial cells via mobilization of antioxidative and DNA repair mechanisms,” Free Radical Biology & Medicine, vol. 52, pp. 2234–2245, 2012. View at: Google Scholar
- J. M. Smoliga, J. A. Baur, and H. A. Hausenblas, “Resveratrol and health—a comprehensive review of human clinical trials,” Molecular Nutrition & Food Research, vol. 55, no. 8, pp. 1129–1141, 2011. View at: Publisher Site | Google Scholar
- H. Ghanim, C. L. Sia, S. Abuaysheh et al., “An antiinflammatory and reactive oxygen species suppressive effects of an extract of Polygonum cuspidatum containing resveratrol,” The Journal of Clinical Endocrinology & Metabolism, vol. 95, no. 9, pp. E1–E8, 2010. View at: Publisher Site | Google Scholar
- H. Ghanim, S. Abuaysheh, C. L. Sia et al., “Increase in plasma endotoxin concentrations and the expression of toll-like receptors and suppressor of cytokine signaling-3 in mononuclear cells after a high-fat, high-carbohydrate meal: implications for insulin resistance,” Diabetes Care, vol. 32, no. 12, pp. 2281–2287, 2009. View at: Publisher Site | Google Scholar
- H. Ghanim, C. L. Sia, K. Korzeniewski et al., “A resveratrol and polyphenol preparation suppresses oxidative and inflammatory stress response to a high-fat, high-carbohydrate meal,” The Journal of Clinical Endocrinology & Metabolism, vol. 96, no. 5, pp. 1409–1414, 2011. View at: Publisher Site | Google Scholar
- P. Brasnyó, G. A. Molnár, M. Mohás et al., “Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients,” The British Journal of Nutrition, vol. 106, no. 3, pp. 383–389, 2011. View at: Google Scholar
- L. M. Vislocky and M. L. Fernandez, “Biomedical effects of grape products,” Nutrition Reviews, vol. 68, no. 11, pp. 656–670, 2010. View at: Publisher Site | Google Scholar
- A. A. A. Bertelli and D. K. Das, “Grapes, wines, resveratrol, and heart health,” Journal of Cardiovascular Pharmacology, vol. 54, no. 6, pp. 468–476, 2009. View at: Publisher Site | Google Scholar
- M. M. Dohadwala and J. A. Vita, “Grapes and cardiovascular disease,” The Journal of Nutrition, vol. 139, no. 9, pp. 17885–17935, 2009. View at: Publisher Site | Google Scholar
- W. R. Leifert and M. Y. Abeywardena, “Grape seed and red wine polyphenol extracts inhibit cellular cholesterol uptake, cell proliferation, and 5-lipoxygenase activity,” Nutrition Research, vol. 28, no. 11, pp. 729–737, 2008. View at: Publisher Site | Google Scholar
- T. Wallerath, G. Deckert, T. Ternes et al., “Resveratrol, a polyphenolic phytoalexin present in red wine, enhances expression and activity of endothelial nitric oxide synthase,” Circulation, vol. 106, no. 13, pp. 1652–1658, 2002. View at: Publisher Site | Google Scholar
- T. Wallerath, H. Li, U. Gödtel-Ambrust, P. M. Schwarz, and U. Förstermann, “A blend of polyphenolic compounds explains the stimulatory effect of red wine on human endothelial NO synthase,” Nitric Oxide, vol. 12, no. 2, pp. 97–104, 2005. View at: Publisher Site | Google Scholar
- J. F. Leikert, T. R. Räthel, P. Wohlfart, V. Cheynier, A. M. Vollmar, and V. M. Dirsch, “Red wine polyphenols enhance endothelial nitric oxide synthase expression and subsequent nitric oxide release from endothelial cells,” Circulation, vol. 106, no. 13, pp. 1614–1617, 2002. View at: Publisher Site | Google Scholar
- P. Gresele, P. Pignatelli, G. Guglielmini et al., “Resveratrol, at concentrations attainable with moderate wine consumption, stimulates human platelet nitric oxide production,” The Journal of Nutrition, vol. 138, no. 9, pp. 1602–1608, 2008. View at: Google Scholar
- D. O. Kennedy, E. L. Wightman, J. L. Reay et al., “Effects of resveratrol on cerebral blood flow variables and cognitive performance in humans: a double-blind, placebo-controlled, crossover investigation,” The American Journal of Clinical Nutrition, vol. 91, no. 6, pp. 1590–1597, 2010. View at: Publisher Site | Google Scholar
- R. H. X. Wong, P. R. C. Howe, J. D. Buckley, A. M. Coates, I. Kunz, and N. M. Berry, “Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure,” Nutrition, Metabolism and Cardiovascular Diseases, vol. 21, no. 11, pp. 851–856, 2011. View at: Publisher Site | Google Scholar
- B. Agarwal and J. A. Baur, “Resveratrol and life extension,” Annals of the New York Academy of Sciences, vol. 1215, no. 1, pp. 138–143, 2011. View at: Publisher Site | Google Scholar
- J. L. Crespo and M. N. Hall, “Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae,” Microbiology and Molecular Biology Reviews, vol. 66, no. 4, pp. 579–591, 2002. View at: Publisher Site | Google Scholar
- I. Bjedov, J. M. Toivonen, F. Kerr et al., “Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster,” Cell Metabolism, vol. 11, no. 1, pp. 35–46, 2010. View at: Publisher Site | Google Scholar
- N. Anisimov, M. A. Zabezhinski, I. G. Popovich et al., “Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice,” Cell Cycle, vol. 10, no. 24, pp. 4230–4236, 2011. View at: Publisher Site | Google Scholar
- S. Majumder, A. Caccamo, D. X. Medina et al., “Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1β and enhancing NMDA signaling,” Aging Cell, vol. 11, no. 2, pp. 326–335, 2012. View at: Publisher Site | Google Scholar
- G. Payne, A. Laporte, R. Deber, and P. C. Coyte, “Counting backward to health care’s future: using time-to-death modeling to identify changes in end-of-life morbidity and the impact of aging on health care expenditures,” The Milbank Quarterly, vol. 85, no. 2, pp. 213–257, 2007. View at: Publisher Site | Google Scholar
- A. Yazdanyar and A. B. Newman, “The burden of cardiovascular disease in the elderly: morbidity, mortality, and costs,” Clinics in Geriatric Medicine, vol. 25, no. 4, pp. 563–577, 2009. View at: Publisher Site | Google Scholar
- J. Mesterton, A. Wimo, Å. By, S. Langworth, B. Winblad, and L. Jönsson, “Cross sectional observational study on the societal costs of Alzheimer’s disease,” Current Alzheimer Research, vol. 7, no. 4, pp. 358–367, 2010. View at: Publisher Site | Google Scholar
- G. Atzmon, C. Schechter, W. Greiner, D. Davidson, G. Rennert, and N. Barzilai, “Clinical phenotype of families with longevity,” Journal of the American Geriatrics Society, vol. 52, no. 2, pp. 274–277, 2004. View at: Publisher Site | Google Scholar
- G. Atzmon, M. Rincon, P. Rabizadeh, and N. Barzilai, “Biological evidence for inheritance of exceptional longevity,” Mechanisms of Ageing and Development, vol. 126, no. 2, pp. 341–345, 2005. View at: Publisher Site | Google Scholar
- N. Barzilai and I. Gabriely, “Genetic studies reveal the role of the endocrine and metabolic systems in aging,” The Journal of Clinical Endocrinology & Metabolism, vol. 95, no. 10, pp. 4493–4500, 2010. View at: Publisher Site | Google Scholar
- M. C. Haigis and B. A. Yankner, “The aging stress response,” Molecular Cell, vol. 40, no. 2, pp. 333–344, 2010. View at: Publisher Site | Google Scholar
- D. Nipič, A. Pirc, B. Banič, D. Šuput, and I. Milisav, “Preapoptotic cell stress response of primary hepatocytes,” Hepatology, vol. 51, no. 6, pp. 2140–2151, 2010. View at: Publisher Site | Google Scholar
Copyright
Copyright © 2012 Samo Ribarič. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.