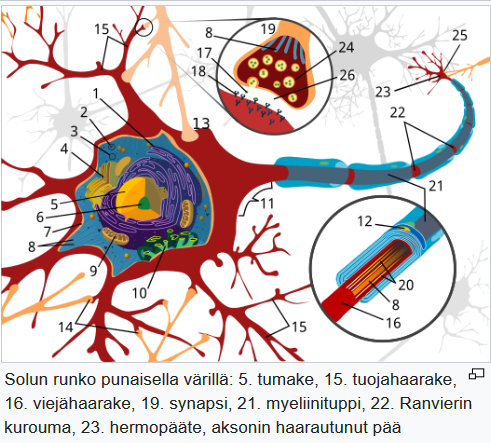
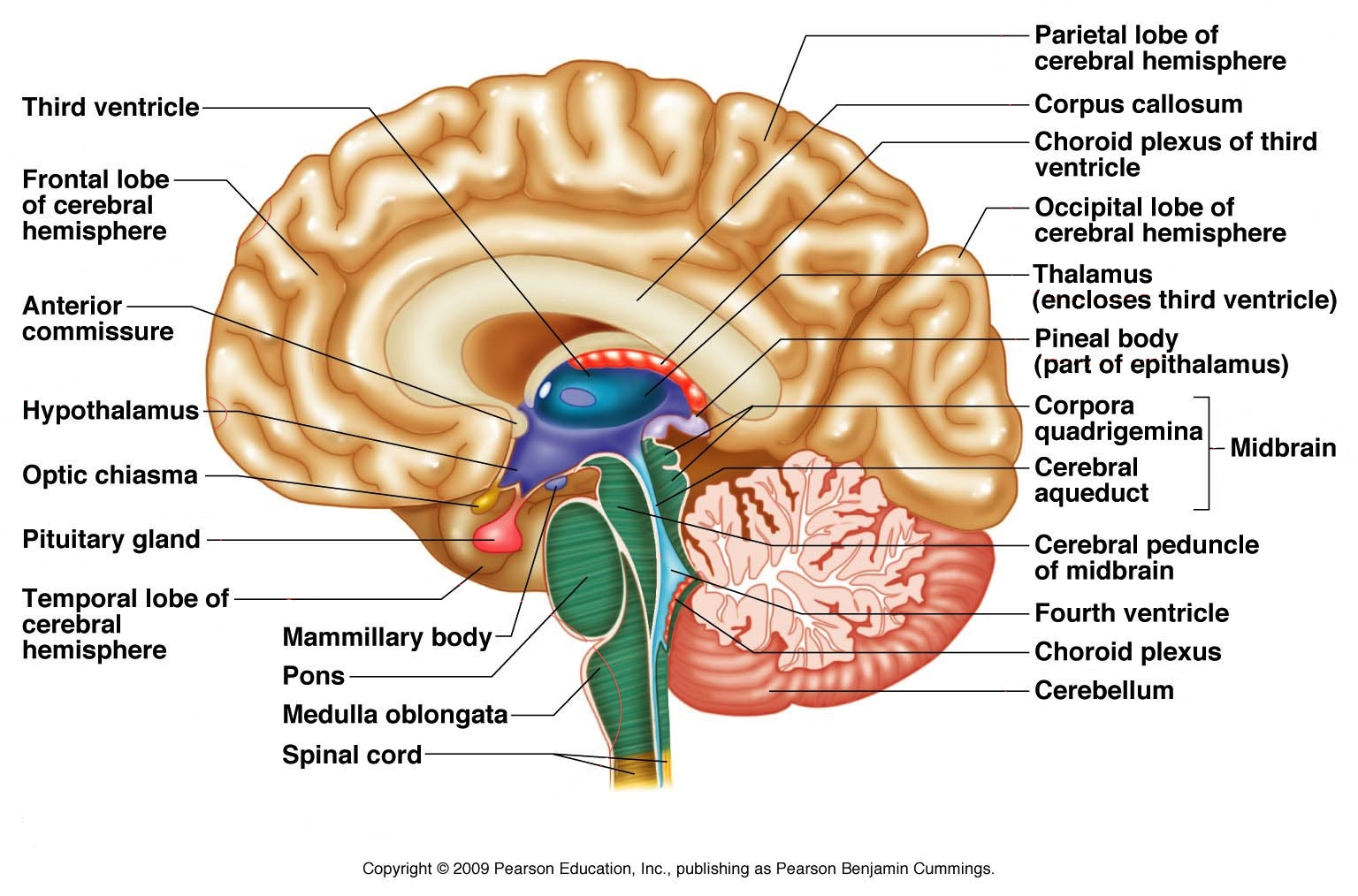
Neuromuskulaariset sairaudet ovat joukko sairauksia, jotka vaikuttavat lihaksia sääteleviin hermoihin tai heikentävät itse lihaksia. Esimerkiksi ALS.
Neurodegeneratiivinen sairaus on yleistermi useille sairauksille, jotka rappeuttavat aivojen ja keskushermoston soluja, kuten neuroneja. Neuronit eivät yleensä uusiudu tai korjaa itseään, joten jos ne surkastuvat ja kuolevat, elimistö ei voi korvata niitä. Neurodegeneratiivisia sairauksista ovat mm. Parkinsonin tauti, Alzheimerin tauti ja Huntingtonin tauti.
Johdanto
Päivä päivältä useammat tutkimukset osoittavat ketogeenisten ruokavalioiden kiistattomat hyödyt metabolisten sairauksien, kuten lihavuuden, metabolisen oireyhtymän ja aikuistyypin diabeteksen hoidossa.
Olen ketoillut puolivallattomasti joulukuun alusta alkaen. Joulukuun 2. päivän ja tämän päivän väliin mahtuu useita horjahduksia olut-, makeis- ja burgeriseikkailuineen. Paino on laskenut ~13 kiloa. Verensokeri pysyy KD:llä optimaalisena ja verenpaine hyvällä tasolla. Oloni on hyvin motivoitunut ja aktiivinen. Arkista huuhailua ei haittaa nälkä tai aivosumu.
Neurologisten häiriöiden osalta ketogeeninen ruokavalio hyväksytään tehokkaaksi terapiaksi farmakoresistentin epilepsian hoidossa, mutta uudet tutkimukset viittaavat siihen, että ketogeeninen ruokavalio voi olla hyödyllinen myös amyotrofisen lateraaliskleroosin (ALS), Alzheimerin- ja Parkinsonin taudin sekä eräiden mitokondriopatioiden yhteydessä.
Esittelin Ruokasodassa aimmin tutkimuskatsauksen, jonka mukaan ketogeeninen ruokavalio voi hidastaa myös multippeliskleroosin etenemistä.
Vaikka näillä sairauksilla on erilaiset patogeneesit, on olemassa eräitä yleisiä mekanismeja, jotka voivat selittää ketogeenisten ruokavalioiden hyötyjä:
- ketogeenisen ruokavalion metaboliset mekanismit tarjoavat tehokkaan energianlähteen sellaisten neurodegeneratiivisten sairauksien hoidossa, joille on tunnusomaista fokaalinen aivojen hypometabolia, eli solujen energiansaannin heikkeneminen
- ketogeeninen ruokavalio vähentää erityyppisiin metabolisiin stresseihin liittyviä oksidatiivisia vaurioita ja inflammaatiota
- ketogeeninen ruokavalio lisää mitokondrioiden biogeneesireittejä
- ketonit ohittavat joihinkin neurologisiin sairauksiin liittyvän kompleksin I aktiivisuuteen liittyvän vian
Tässä katsauksessa tutustutaan ketogeenisen ruokavalion neuroprotektiivisiin aineenvaihduntamekanismeihin
Yksittäisillä ravintoaineilla voi olla positiivisia vaikutuksia luurankolihasten terveyteen. Lisäksi ravintoaineiden yhdistelmät voivat vaimentaa joidenkin hermo-lihassairauksien oireita. Toisaalta tiedetään myös, että laihduttamisen vaikutukset terveyteen liittyvät eri makro- ja hivenravinteiden keskinäiseen saantisuhteeseen, eikä niinkään yksittäisin ravintoaineeseen.
Ketogeeninen ruokavalio (KD) on viime vuosina herättänyt suurta kiinnostusta. 1900-luvun kolmannelta vuosikymmeneltä lähtien ketogeenistä ruokavaliota on käytetty farmakologisesti resistentin epilepsian hoitoon [1–3]. Viime aikoina KD:n on havaittu olevan toimiva terapia myös monissa täysin erilaisissa sairauksissa, kuten lihavuus [4], PCOS [5], syöpä [1, 6, 7], diabetes [8] ja muut patologiset tilat [9– 11].
Vaikka monet tutkimukset ovat osoittaneet KD:n potentiaaliset positiiviset vaikutukset moniin neurologisiin ja hermo-lihassairauksiin, vain harvat tutkimukset ovat tutkineet tämän lupaavan ravitsemuksellisen lähestymistavan mekanismeja [12].
Tämän katsauksen tarkoituksena on tarkastella KD:n roolia hermostoon ja lihasten toimintaan vaikuttavissa sairauksissa.

Ketogeenisessä ruokavaliossa
Muutaman päivän paastoamisen tai hiilihydraattien huomattavan rajoittamisen (alle 20 g päivässä) seurauksena glukoosivarastot eivät riitä:
- normaaliin rasvan hapettumiseen oksaloasetaatin syöttämisen kautta sitruunahappokierrossa (Krebsin syklissä, TCA-sykli) ja
- keskushermoston glukoositarpeen tyydyttämiseen [13, 14] (kuva 1)
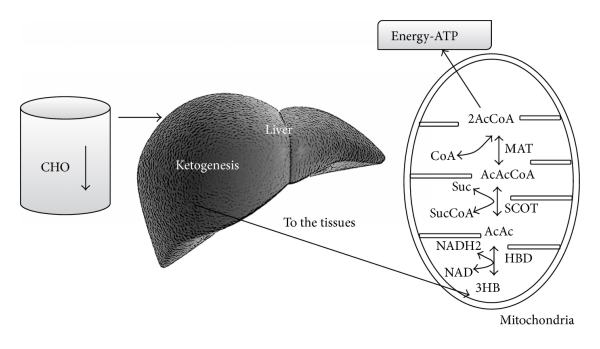 Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.
Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.
Oksaloasetaatin määrän tulisi pysyä tasolla, joka on riittävä sitruunahappokierron toiminnan (ts. asetyyli-CoA:n ja oksaloasetaatin välinen kondensaatio) mahdollistamiseksi.
Oksaloasetaatti on epävakaa ja se on uudelleenkoottava (tällaisia reaktioita kutsutaan anaplerooteiksi). Elimistölle helpoin tapa tuottaa oksaloasetaattia on pyruvaatista, joka saadaan glukoosista.
| Oksaloetikkahappo (oksaloasetaatti) on ketodikarboksyylihappoihin kuuluva orgaaninen yhdiste. Oksaloetikkahappo on välituote useissa biokemiallisesti tärkeissä reaktioissa.
Sitruunahappokierron ensimmäisessä vaiheessa oksaloetikkahappo ja asetyylikoentsyymi-A reagoivat sitraattisyntaasientsyymin katalysoimana muodostaen sitruunahappoa. Reaktiossa asetyyliryhmä siirretään oksaloetikkahapon ketonihiileen ja karbonyyliryhmä pelkistetään hydroksyyliryhmäksi.
Oksaloetikkahappoa muodostuu sitruunahappokierron kymmenennessä vaiheessa, kun omenahappo dehydrataan malaattidehydrogenaasientsyymin avulla.
Glukoneogeneesin ensimmäisessä vaiheessa palorypälehappo muutetaan oksaloetikkahapoksi. Reaktio kuluttaa ATP-molekyylin ja sitä katalysoi pyruvaattikarboksylaasientsyymi.
Glukoneogeneesin toisessa vaiheessa oksaloetikkahappo muutetaan fosfoenolipalorypälehapoksi. Reaktiossa tarvittava energia saadaan hydrolysoimalla yksi gyanosiinitrifosfaatin korkeaenergiainen fosfaattiryhmä. Reaktiossa muodostuu lisäksi hiilidioksidia ja guanosiinidifosfaattia. Reaktiota katalysoi fosfoenolipyruvaattikarboksikinaasi.
Eliöt voivat tuottaa aminohappo asparagiinihappoa oksaloetikkahaposta. Asparagiinihappoa syntyy, kun oksaloetikkahappo ja glutamiinihappo reagoivat. Reaktio on transaminaatioreaktio ja sitä katalysoi eräs transaminaasien luokkaan kuuluva entsyymi, aspartaattiaminotransferaasi. Koentsyyminä toimii pyridoksaalifosfaatti. – Wikipedia |
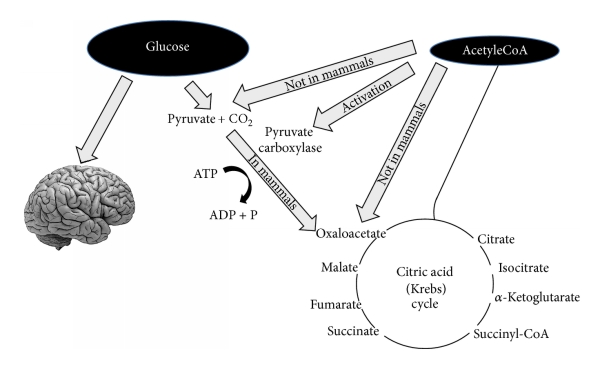
Nisäkkäillä pyruvaattia ei voida tuottaa asetyylikoentsyymi-A:sta, kuten kuvassa on esitetty.
(1) Oksaloasetaatti on ruumiin normaalilämmössä melko epävakaa molekyyli. Sitä ei voi varastoida mitokondrioiden matriksiin. Tässä ”glukoosinpuutos” -tilassa tarvitaan oksaloasetaattia trikarboksyylihapposyklin tehokkaan toiminnan varmistamiseksi. Oksaloasetaatti toimitetaan anapleroottisyklin kautta, joka syntetisoi oksaloasetaatin glukoosista pyruviinihapon ATP-riippuvaisen karboksyloinnin kautta pyruvaattikarboksylaasilla [15].
(2) Koska keskushermosto ei voi käyttää vapaita rasvahappoja (FFA) betaoksidaatiossa suoraan energialähteenä (vapaat rasvahapot eivät läpäise veri-aivoestettä), keskushermosto käyttää yleensä solujen energiasubstraattina glukoosia.
3-4 päivän kuluttua hiilihydraattien saannin rajoittamisesta, keskushermoston on löydettävä soluille vaihtoehtoinen energialähde, kuten Felig et al. [13, 14, 16, 17] ovat osoittaneet. Vaihtoehtoisen energianlähteen soluille tarjoavat maksan tuottamat ketoaineet (KB): asetoasetaatti (AcAc), 3-hydroksibutyraatti (3HB, beta-hydroksibutyraatti) ja asetoni [18], joita saadaan asetyylikoentsyymi-A:n ylituotannosta ilman oksaloasetaatin samanaikaista tuotantoa. Energiasubstraateiksi kelpaavia ketoaineita sytyy ketogeneesissä. Ketogeneesiä tapahtuu pääasiassa maksan mitokondrioiden matriksissa [19].
Maksa tuottaa ketoaineita, mutta ei pysty käyttämään niitä, koska maksassa ei ole sukkinyyli-CoA: 3-CoA transferaasi (SCOT) -entsyymiä, jota tarvitaan asetoasetaatin muuttamiseksi asetyylikoentsyymi-A:ksi [18]. maksan ketogeneesissä syntetisoidaan asetoasetaattia, mutta ensisijainen verenkierrossa kiertävä ketoaine on 3-hydroksibutyraatti, eli beta-hydroksibutyraatti.
Normaaleissa olosuhteissa vapaan asetoasetaatin tuotanto on vähäistä ja se voi metaboloitua useissa kudoksissa, kuten luurankolihaksessa ja sydämessä. Asetoasetaatin ylituotanto-olosuhteissa sitä syntyy normaalia enemmän, mutta osa siitä syntetisoidaan kahdeksi muuksi ketoaineeksi.
Korkea ketoaineiden taso veressä ja niiden eliminaatio virtsan kautta aiheuttaa ketonemiaa ja ketonuriaa. Normaaleissa olosuhteissa ketoaineiden pitoisuus on yleensä hyvin matala (<0,3 mmol / l) verrattuna glukoosiin (noin 4-5 mmol / l) [20, 21].
Kun ketoaineet ovat saavuttaneet noin 4 mmol / l konsentraation, keskushermosto alkaa käyttää niitä energialähteenä [21]. Kudokset käyttävät ketoaineita energialähteenä [19, 21, 23] aineenvaihduntareitin kautta, joka muuntaa ensin beta-hydroksibutyraatin (3HB) takaisin asetyyliasetaatiksi, joka sitten muutetaan asetoasetyylikoentsyymi-A:ksi. Jälkimmäinen jaetaan lopulta kahteen asetyylkoentsyymi-A-molekyyliksi, joita käytetään myöhemmin sitruunahappokierrossa (kuva 2).
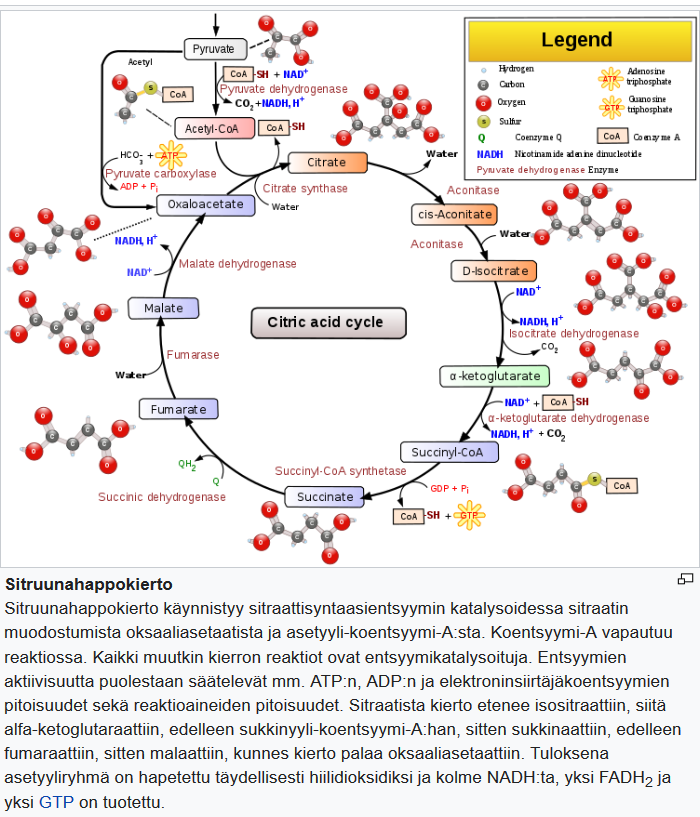
On mielenkiintoista huomata, että verensokeriin verrattuna ketoaineet pystyvät tuottamaan suuremman energiamäärän johtuen niiden aiheuttamista mitokondrioiden ATP-tuotannon muutoksista [21, 24, 25].
Ruokavalion hiilihydraattien vähentynyt saatavuus johtaa lisääntyneeseen ketoaineiden tuotantoon maksassa. Maksa ei voi käyttää ketoaineita, koska siitä puuttuu mitokondrioiden tarvitsema entsyymi sukkinyyli-CoA: 3-ketohappo (oksohappo) CoA-transferaasi (SCOT), jota tarvitaan asetoasetaatin aktivaatioon asetoasetyyli-CoA:ksi. Ketoaineita hyödyntävät kudokset, erityisesti aivot. Ketoaineet siirtyvät sitruunahappokiertoon muututtuaan asetyylikoentsyymi-A:ksi sukkinyyli-CoA: 3-CoA-transferaasin (SCOT) ja metyyliasetoasetyyli-CoA-tiolaasin (MAT) avulla. Vaikka glukoosi on vähentynyt, se pysyy fysiologisilla tasoilla [26, 27] johtuen kahdesta päälähteestä:
(1) glukogeenisista aminohapoista ja
(2) triglyserideistä hajotuksen kautta vapautuneesta glyserolista [28, 29]
Fysiologisen ketoosin (nopea tai erittäin vähäkalorinen ketogeeninen ruokavalio) aikana ketonemia saavuttaa maksimiarvot 7/8 mmol / l ilman pH:n muutoksia, kun taas hallitsemattomassa diabeettisessa ketoasidoosissa ketoaineiden pitoisuus voi ylittää 20 mmol / l, kun veren pH laskee samanaikaisesti.
 Terveiden ihmisten veren ketoaineiden pitoisuudet eivät ylitä 8 mmol / l, koska keskushermosto (CNS) käyttää näitä molekyylejä tehokkaasti energiasubstraatteina glukoosin sijasta [16]. Taulukossa veren ketoaineiden tasot normaalin ruokavalion ja ketogeenisen ruokavalion aikana (ts. 20 grammaa hiilihydraatteja päivässä) sekä diabeettinen ketoasidoosi [10] .
Terveiden ihmisten veren ketoaineiden pitoisuudet eivät ylitä 8 mmol / l, koska keskushermosto (CNS) käyttää näitä molekyylejä tehokkaasti energiasubstraatteina glukoosin sijasta [16]. Taulukossa veren ketoaineiden tasot normaalin ruokavalion ja ketogeenisen ruokavalion aikana (ts. 20 grammaa hiilihydraatteja päivässä) sekä diabeettinen ketoasidoosi [10] .
Ketogeeninen ruokavalio imitoi paastoa ja sen aineenvaihduntamekanismeja
Perinteisesti lääkärit vierastavat ketoosia, koska he yhdistävät ketoosin insuliinin puutteesta johtuvaan vaikeaan hyperketonemiaan, joka voi aiheuttaa vakavan ketoasidoosin ja kuoleman tyypin 1 diabetesta sairastavilla henkilöillä.
Hans Krebs oli ensimmäinen, joka käytti termiä ”fysiologinen ketoosi” erottaakseen paaston ja ketogeenisen ruokavalion aiheuttaman lievän (8 mmol / l ketonipitoisuuden) ketoosin metabolisesti epätasapainoisen diabeteksen ”patologisesta ketoasidoosista” [31].
Paaston tai aliravitsemuksen jaksot ovat siinä mielessä ketogeenisiä [23], että insuliinin ja glukoosin pitoisuudet laskevat, kun taas glukagonin pitoisuudet kasvavat normaalia verensokeritason ylläpitämiseksi. Kun keho siirtyy ravinnon runsauden tilasta ravinnon puutteen tilaan (ketoosiin esimerkiksi ketogeenisen ruokavalion simuloidun ravinnon puutteen kautta), veren vapaiden rasvahappojen (FFA) ja ketoaineiden pitoisuus kasvaa pienellä viiveellä.
Tästä näkökulmasta tarkasteltuna ketogeenistä ruokavaliota voidaan verrata kalorirajoitukseen, aliravitsemukseen tai paastoamiseen. Tämä ravinteiden saannin metabolisen vasteen manipulointi, sekä määrällisesti että laadullisesti, vaikuttaa sekä verensokeriin että ketoaineisiin. Sillä on myös kyky edistää aineenvaihduntareittien ja soluprosessien, kuten stressiresistenssin ja autofagian muutoksia.
Ketogeeniset ruokavaliot voivat toimia myös samalla tavalla kuin kalorirajoitus (CR) AMPK:lla ja SIRT-1:llä [33]. Jotta ymmärtäisimme ketogeenisen ruokavalion monimutkaiset vaikutukset ja mekanismit elimistössä, meidän on otettava huomioon nämä solunsisäiset molekyylitason aineenvaihduntareitit.
PGC1α aktivoituu fosforyloidussa tilassa. Fosforyloitu PGC1α siirtyy sytosolista solun tumaan, jossa se edistää rasvahappojen kuljetusta, rasvan hapettumista ja oksidatiivista fosforylaatiota sekä osallistuvien geenien transkriptiota [34].
PGC1α voidaan fosforyloida useiden eri aineenvaihduntareittien kautta, kuten AMPK, kalsium-kalmoduliinista riippuvainen proteiinikinaasi ja p38-mitogeeniaktivoitunut proteiinikinaasireitti [35]. PGC1α voidaan aktivoida myös SIRT1-välitteisellä deasetylaatiolla [36]. AMPK voi toimia joko PGC1α:n fosforyloimalla tai suoraan.
AMPK-aktivointi estää myös mTOR-signaloinnin. Vaikuttaa kuitenkin epäjohdonmukaiselta estää tärkeä kasvuvälitteinen reitti (mTOR), joka säätelee lihasmassaa, jotta luurankolihakset voivat kasvaa.
Ravinteiden manipulointi vaikuttaa näihin aineenvaihduntareitteihin; esim. hiilihydraattien puutteellinen saanti stimuloi in vivo AMPK:n ja SIRT-1:n aktivaatiota, lisäämällä AMPK:n fosforylaatiota ja PGC1α:n deasetylointia luurankolihaksissa vaikuttamatta AMPK:n, PGC1α:n tai SIRT 1: n kokonaismäärään.
Nämä mekanismit näyttävät aktivoituvan muutaman tunnin (5 tunnin) paaston jälkeen hiirillä [39]. Aktivoiduttuaan SIRT1 ja AMPK tuottavat hyödyllisiä vaikutuksia glukoosin homeostaasissa ja insuliinimetaboliassa [40].
Askel vaikeampaan: mikä helvetin AMPK?
|
Tämä on teknisempää settiä. 5′-AMP-aktivoitu proteiinikinaasi, AMPK tai 5′-adenosiinimonofosfaatilla aktivoitu proteiinikinaasi on entsyymi, jolla on suuri vaikutus soluenergian homeostaasissa.
AMPK vaikuttaa erityisesti aktivoimalla glukoosin ja rasvahappojen imeytymistä ja hapettumista, kun solujen energiataso on matala. AMPK:ta ei tule sekoittaa cAMP-aktivoituun proteiinikinaasiin.
AMPK koostuu kolmesta proteiinista (α, β, γ -alayksiköistä), jotka yhdessä muodostavat toiminnallisen entsyymin. AMPK ilmentyy useissa kudoksissa, kuten maksassa, aivoissa ja luurankolihaksissa.
AMPK-aktivaation nettovaikutus vasteena AMP:n ja ADP:n sitoutumiselle on maksan rasvahappojen hapettumisen stimulointi, ketogeneesi, luustolihasten rasvahappojen hapettumisen stimulointi ja glukoosin imeytymisen tehostaminen, kolesterolisynteesin esto, lipogeneesin ja triglyseridisynteesin esto sekä, rasvasolujen lipogeneesin ja lipolyysin esto ja insuliinin erityksen modulointi haiman beetasoluista. |
| Kullakin AMPK:n kolmella alayksiköllää on erityinen rooli AMPK:n vakaudessa ja aktiivisuudessa. Vaikka AMPK:N yleisimmät isoformit, jotka ilmentyvät useimmissa soluissa, ovat α1-, β1- ja γ1-isoformeja, on osoitettu, että α2-, β2-, γ2- ja γ3-isoformit ilmentyvät myös sydämen ja luuston lihaksissa. |
| Johtuen AMPK:n komponenttien isoformeista, nisäkkäillä on 12 versiota AMPK:sta, joista jokaisella voi olla erilainen kudospaikannus ja erilaiset toiminnot eri olosuhteissa. AMPK:ta säännellään allosterisesti ja translaation jälkeisillä muokkauksilla, jotka toimivat yhdessä. Jos AMPK:n alfa-alayksikön tähde T172 fosforyloidaan, AMPK aktivoituu; fosfataasien pääsy tähän jäännökseen estetään, jos AMP tai ADP voivat estää pääsyn ja ATP voi syrjäyttää AMP:n ja ADP:n.
AMPK:ta säätelee allosterisesti enimmäkseen kilpaileva sitoutuminen gamma-alayksikköön ATP:n (joka sallii fosfataasipääsyn T172: een) ja AMP:n tai ADP:n (joista kukin estää pääsyn fosfataaseihin) välillä.
Näin ollen näyttää siltä, että AMPK on AMP / ATP- tai ADP / ATP-suhteiden ja siten solujen energiatason anturi.
On eräitä aineenvaihduntamekanismeja, joilla insuliini, leptiini ja diasyyliglyseroli estävät AMPK: ta indusoimalla erilaisia muita fosforylaatioita. AMPK:ta voidaan estää tai aktivoida erilaisilla kudosspesifisillä ubikitinaatioilla. Sitä säätelevät myös useat proteiini-proteiini-vuorovaikutukset, ja hapettavat tekijät voivat joko aktivoida tai estää niitä. Kun AMPK fosforyloi asetyyli-CoA-karboksylaasi 1: tä (ACC1) tai sterolia säätelevää elementtiä sitovaa proteiinia 1c (SREBP1c), se estää rasvahappojen, kolesterolin ja triglyseridien synteesiä ja aktivoi rasvahappojen saannin ja β-oksidaation. AMPK stimuloi glukoosinottoa luurankolihassa fosforyloimalla Rab-GTPaasia aktivoivan proteiinin TBC1D1, joka lopulta indusoi GLUT1-rakkuloiden fuusion plasmamembraaniin. AMPK stimuloi glykolyysiä aktivoimalla 6-fosfofrukto-2-kinaasi / fruktoosi-2,6-bisfosfataasi 2/3: n fosforylaation ja aktivoimalla glykogeenifosforylaasin fosforylaation, ja se estää glykogeenisynteesiä estävän fosforyylin kautta
Monet luurankolihasten biokemialliset muutokset, jotka tapahtuvat yhden harjoittelun tai pitkittyneen harjoittelun aikana, kuten lisääntynyt mitokondrioiden biogeneesi ja kapasiteetti, lisääntynyt lihasten glykogeeni ja lisääntynyt entsyymien erikoistuminen glukoosinottoon soluissa, kuten GLUT4 ja heksokinaasi II, ovat todennäköisesti ainakin osittain AMPK:n aktivaation välittämiä tapahtumia. |
| AMPK:lla on keskeinen rooli liikunnan / treenattujen lihassolujen verenkierron lisäämisessä stimuloimalla ja vakauttamalla sekä vaskulogeneesiä että angiogeneesiä.
Yhdessä nämä muutokset ilmenevät todennäköisesti sekä väliaikaisen että ylläpidetyn AMPK-aktiivisuuden lisääntymisen seurauksena, jonka AMP : ATP-suhteen nousu aiheuttaa yksittäisten liikuntajaksojen ja pitkäaikaisen harjoittelun aikana.
Yhden akuutin harjoittelun aikana AMPK antaa supistuvien lihassolujen sopeutua energiahaasteisiin lisäämällä heksokinaasi II: n ilmentymistä, GLUT4:n translokaatiota plasmakalvoon, glukoosinottoa ja stimuloimalla glykolyysiä. Jos liikunta jatkuu pitkitettynä harjoituksena, AMPK ja muut signaalit helpottavat lihasten adaptoitumista sopeuttamalla lihassolujen aktiivisuuden aineenvaihdunnan muutokseen, mikä johtaa rasvahappojen hapettumisen kautta muodostuvaaan ATP:hen glykolyysin sijaan.
AMPK saavuttaa siirtymisen oksidatiiviseen metaboliaan säätelemällä ja aktivoimalla oksidatiivisia entsyymejä, kuten heksokinaasi II, PPARalpha, PPARdelta, PGC-1, UCP-3, sytokromi C ja TFAM. AMPK-aktiivisuus kasvaa liikunnan seurauksena ja LKB1 / MO25 / STRAD-kompleksia pidetään 5′-AMP-aktivoidun proteiinikinaasin tärkeimpänä ylävirran AMPKK:na. Tämä on hämmentävä ilmiö, kun otetaan huomioon, että vaikka AMPK-proteiinien määrä kasvaa luukudoksessa kestävyysharjoittelun vaikutuksesta, niiden aktiivisuus vastaavasti laskee kestävyysharjoittelun seurauksena.
On mahdollista, että on olemassa suora yhteys kestävyysharjoitettujen luurankolihasten havaitun AMPK-aktiivisuuden vähenemisen ja kestävyysharjoitteluun liittyvän AMPK-vasteen ilmeisen vähenemisen välillä. |
| Yksi keskeisistä reiteistä AMPK:n rasvahappojen hapettumisen säätelyssä on asetyyli-CoA-karboksylaasin fosforylaatio ja inaktivaatio. Asetyyli-CoA-karboksylaasi (ACC) muuntaa asetyyli-CoA n malonyyli-CoA:ksi, joka on karnitiinipalmmityylitransferaasi 1:n (CPT-1) estäjä. CPT-1 kuljettaa rasvahapot mitokondrioihin hapetusta varten. ACC: n inaktivointi johtaa siis lisääntyneeseen rasvahappokuljetukseen ja sitä seuraavaan hapettumiseen.
Todennäköisesti malonyyli-CoA:n väheneminen tapahtuu malonyyli-CoA-dekarboksylaasin (MCD) vaikutuksesta, jota AMPK voi säätää. MCD on ACC:n antagonisti dekarboksyloimalla malonyyli-CoA:ta asetyyli-CoA:ksi, mikä johtaa vähentyneeseen malonyyli-CoA:han ja lisääntyneeseen CPT-1:een ja rasvahappojen hapettumiseen.
AMPK: lla on myös tärkeä rooli maksan lipidien aineenvaihdunnassa. On pitkään tiedetty, että maksan ACC:tä on säännelty maksassa fosforylaatiolla. AMPK fosforyloi ja inaktivoi myös 3-hydroksi-3-metyyliglutaryyli-CoA-reduktaasin (HMGCR), joka on kolesterolisynteesin avainentsyymi. HMGR muuntaa 3-hydroksi-3-metyyliglutaryyli-CoA:n, joka on valmistettu asetyylikoentsyymi-A:sta, mevalonihapoksi, joka sitten kulkee useita muita metabolisia vaiheita kolesteroliksi.
AMPK auttaa säätelemään rasvahappojen hapettumista ja kolesterolisynteesiä. Insuliini on hormoni, joka auttaa säätelemään elimistön glukoosipitoisuutta. Kun verensokeri on korkea, insuliinia vapautuu Langerhansin saarekkeiden beetasoluista. Insuliini muun muassa helpottaa glukoosin imeytymistä soluihin lisäämällä glukoosinkuljettaja GLUT-4:n ilmentymistä ja siirtymistä.
AMPK ja kilpirauhashormoni säätelevät eräitä samanlaisia prosesseja. Nämä yhtäläisyydet tuntien Winder ja Hardie et al. suunnittelivat kokeen selvittääkseen, vaikuttiko kilpirauhashormoni AMPK: hon. He havaitsivat, että kaikki AMPK:n alayksiköt lisääntyivät luurankolihaksissa kilpirauhashormonihoidolla. Myös fosfo-ACC: n, AMPK-aktiivisuuden markkerin, määrä lisääntyi.
AMPK:n häviämisen on raportoitu muuttavan glukoosia tunnistavien solujen herkkyyttä huonosti tunnettujen mekanismien kautta. AMPKα2-alayksikön menetys haiman beetasoluissa ja hypotalamuksen neuroneissa vähentää näiden solujen herkkyyttä solunulkoisen glukoosipitoisuuden muutoksille.
Altistuminen toistuville insuliinin aiheuttamille hypoglykemioille laski AMPK:n aktivaatiota hypotalamuksessa ja samalla esti hypoglykemian vasta-ainevasteen rottakokeissa.
AMPK säätyy lysosomeissa useiden kliinisesti merkittävien järjestelmien kautta. Näihin sisältyy AXIN – LKB1 -kompleksi, joka toimii vasteena AMP-tunnistuksesta riippumatta toimiviin glukoosirajoituksiin, joka tunnistavat matalan glukoosin fruktoosi-1,6-bisfosfaatin puuttumisena dynaamisen vuorovaikutussarjan kautta kosketuksissa olevien lysosomaalisesti lokalisoidun V-ATPaasi-aldolaasin välillä.
Toinen lysosomeihin lokalisoitu AMPK-ohjausjärjestelmä riippuu Galectin-9-TAK1-järjestelmästä ja ubikvitinaatiovasteista kontrolloiduilla deubikitinoivilla entsyymeillä, kuten USP9X, mikä johtaa AMPK:n aktivaatioon vasteena lysosomaalisille vaurioille.
Nämä lysosomaaliset vauriot voivat ilmentyä biokemiallisesti ja fyysisesti proteiiniaggregaattien, kuten proteopaattisten tau-proteiinien kautta Alzheimerin taudissa, kiteisinä piidioksideina, jotka aiheuttavat silikoosia, kolesterolikiteinä, jotka aiheuttavat inflammaatiota NLRP3-tulehduksen kautta ja kihtiin liittyvinä uraattikiteinä, tai mikrobien hyökkäyksen aikana, kuten Mycobacterium tuberculosis-infektiossa ja koronavirus-infektiossa.
[62] Molemmat yllä mainitut lysosomaalisesti lokalisoidut AMPK:ta säätelevät järjestelmät aktivoivat vasteena metformiinille, laajalti määrätylle diabeteslääkkeelle. Jotkut todisteet osoittavat, että AMPK:lla voi olla rooli kasvaimen tukahduttamisessa. Tutkimukset ovat osoittaneet, että AMPK voi käyttää suurinta osaa tai jopa kaikkia maksakinaasi B1:n (LKB1) tuumoria tukahduttavista ominaisuuksista. Lisäksi tutkimuksissa, joissa AMPK-aktivaattori metformiinia käytettiin diabeteksen hoitoon, havaittiin korrelaatio vähentyneeseen syöpäriskiin verrattuna muihin lääkkeisiin.
Hiirillä, joilla ei ollut AMPK:ta ilmentävää geeniä, oli kasvanut riski lymfoomien kehittymiselle. Toisaalta jotkut tutkimukset ovat tulkinneet AMPK:n kasvaimen promoottorina, joka suojaa pahanlaatuisia syöpäsoluja. Vaikuttaa siltä, että AMPK voi kääntää takkia syöpäsolujen kohdalla. Kun syöpäsolut ovat muodostuneet organismiin, AMPK alkaakin suojaamaan organismin solujen sijaan pahanlaatuisia syöpäsoluja.
Ei ole suoraa näyttöä siitä, että AMPK:n estäminen olisi tehokas syöpähoito ihmisillä. AMPK:n näennäisesti paradoksaalinen rooli syövän puolustajana/promoottorina toteutuu, kun tarkastelemme lähemmin energiaa tunnistavaa entsyymiä suhteessa liikuntaan ja pitkäaikaiseen harjoitteluun.
Lyhytaikaisen akuutin harjoitusasteikon tavoin pitkäaikaiset kestävyysharjoittelututkimukset paljastavat myös oksidatiivisten metabolisten entsyymien, GLUT-4:n, mitokondrioiden koon ja määrän lisääntymisen ja lisääntyneen riippuvuuden rasvahappojen hapettumisesta. – Wikipedia |

4. Ketogeeninen ruokavalio ja amyotrofinen lateraaliskleroosi
| Amyotrofinen lateraaliskleroosi, ALS ( sclerosis lateralis amyotrophica) eli Lou Gehrigin tauti tai motoneuronitauti (engl. Motor neuron disease, MND) on etenevä motoneuronisairaus, joka rappeuttaa sekä ylempiä että alempia liikehermoja.
ALS aiheuttaa etenevää rappeutumista liikehermosoluissa, jotka ohjaavat tahdosta riippuvien lihasten toimintaa. ALS:n pääasiallisia oireita ovat lihasten jäykkyys, lihasnykäykset ja asteittain pahentuva lihasten heikentyminen niiden koon pienentyessä.
Tämä johtaa vaikeuksiin puhumisessa, nielemisessä, liikkumisessa ja lopulta hengittämisessä. Sairaus ei vaikuta näköön tai kuuloon eikä haju-, maku- tai tuntoaisteihin. Myös sydän, verenkierto sekä ruoansulatus ja eritysjärjestelmät säilyvät vahingoittumattomina.
Kun sairaus on pitkälle edennyt, potilas ei kykene tekemään juuri mitään ilman ulkopuolista apua; jossain vaiheessa joudutaan tukeutumaan hengityskoneeseen, tarjoamaan potilaalle pillillä imettävää nestemäistä ruokaa tai antamaan ruoka PEG-letkun kautta ja auttamaan tätä kaikissa arkipäivän toimissa. Kuoleman aiheuttaa hengityslihasten heikkous.
Sairaus on harvinainen ja parantumaton. Sairauden aiheuttajaa ei toistaiseksi tunneta. Sairausprosessia on todennäköisesti käynnistämässä usean tekijän vuorovaikutus. – Wikipedia |
ALS
Amyotrofinen lateraaliskleroosi (ALS) on progressiivinen neurodegeneratiivinen sairaus, joka vaikuttaa selkärangan ja aivokuoren motorisiin hermosoluihin, mikä johtaa lihasten progressiiviseen heikkenemiseen ja luurankolihasten toimintakyvyn menetykseen.
Tautiin sairastuvat henkilöt kuolevat keskimäärin 2–5 vuoden kuluessa oireiden ilmaantumisesta. Kuolema tapahtuu yleensä hengityshalvauksesta. Tällä hetkellä ALS:lle ei tunneta tehokasta hoitoa.
Ainoa Yhdysvaltain FDA:n hyväksymä farmakologinen hoito rajoittuu rilutsoliin, joka vaikuttaa vain vähän taudin etenemisen ja pidentää elinaikaa vain 2-3 kuukaudella [41]. ALS:n syyt ovat monimutkaisia ja monitekijäisiä. Niihin sisältyy geneettiset tekijät ja ympäristömuuttujat:
- elimistön oksidatiiviset vauriot,
- neurofilamenttien kertyminen soluihin,
- eksitotoksisuus, joka liittyy glutamaattimetabolian häiriöön ja
- mitokondrioiden kalvojen toimintahäiriöt
ovat eräitä oletettuja taudin kehittymiseen vaikuttavia tekijöitä [42–44]. Muiden hermosoluja rappeuttavien neurodegeneratiivisten häiriöiden tapaan mitokondrioiden todennäköinen vaikutus ALS:n patogeneesiin tekee ketogeenisestä ruokavaliosta lupaavan synergisen työkalun ALS: n hoitoon [45].
Mitokondrioiden yhteys
Noin 10%:lla ALS-potilaista sairaus on perinnöllinen (familiaalinen amyotrofinen skleroosi FALS) ja joka viidennellä FALSia sairastavalla on mutaatio geenissä, joka koodaa entsyymiä Cu / Zn superoksididismutaasi 1 (SOD1) [46]. Tämä mutaatio liittyy mitokondrioiden aktiivisuuteen; itse asiassa juuri mutantti SOD1 on lokalisoitu mitokondrioita sitovaan bcl2:een (solun antiapoptoottinen proteiini) [47].
Lisäksi mitokondrioiden kompleksin I aktiivisuuden heikkeneminen on mitattu ALS-potilaiden luurankolihaksissa ja selkäytimissä [48]. Tulokset osoittavat, että ketoaineet voivat vaikuttaa mitokondrioiden toimintaan palauttamalla esimerkiksi kompleksin I toiminnan farmakologisen eston jälkeen. Lisäksi viljellyissä hermosoluissa, joita hoidetaan farmakologisilla aineilla, jotka estävät kompleksin I, ketoaineiden lisäys palauttaa kompleksin toiminnan [49].
Tutkijat osoittivat hiljattain, että ALS-hiirimallissa ketogeeninen ruokavalio johti korkeampaan motoristen hermosolujen eloonjäämiseen ja parempaan motoriseen toimintaan verrattuna hiiriin, jotka eivät saaneet KD-ruokaa [50]. Tutkimuksissa on myös havaittu, että ketoaineiden lisäys (kapryylihappo) paransi mitokondrioiden toimintaa ja motoristen hermosolujen määrää ALS-hiirimallissa [51].
Tutkijat selittivät nämä tulokset DHB:n neuroprotektiivisella vaikutuksella. Lisäksi he arvelivat, että hyperketonemia saattaa parantaa mitokondrioiden vikoja lisäämällä mitokondrioiden energian ja ATP-molekyylien tuotantoa (mitattuna puhdistetuissa mitokondrioissa ALS-hiirimallista). On huomattava, että kummassakaan tutkimuksessa hiirien eloonjääminen ei lisääntynyt merkittävästi.
Ketogeenisen ruokavalion aikana ruokavalion rasvojen prosenttiosuus oli erittäin korkea (60%). Tämä voi selittää energia-aineenvaihdunnan havaitut ja mitatut parannukset.
Kolesteroli ja fosfolipidit ovat välttämättömiä aksonikalvojen terveydelle ja perifeeristen hermokalvojen vamman korjauksille. Erityisen tärkeitä ovat pienitiheyksiset lipoproteiinit [52].
Eräät epidemiologiset havainnot viittaavat siihen, että hyperlipidemia on merkittävä ALS-potilaiden elinajanodotetta lisäävä tekijä [53]. Paganoni et al. [54], osoitti kuitenkin UI-muotoisen yhteyden painoindeksin ja kuolleisuuden välillä, sekä suuremman eloonjäämistodennäköisyyden potilailla, joilla oli korkeampi painoindeksi (BMI: 30–35). Tässä tutkimuksessa dyslipidemia ei osoittanut riippumattomasti potilaan parempaa ennustetta.
Wills et al. [55] osoittivat äskettäin, että potilailla, jotka saivat runsaasti energiaa / runsaasti hiilihydraatteja sisältävää ravintoa, oli vähemmän haittatapahtumia ja kuolemantapauksia kuin runsaasti rasvaa / paljon kaloreita sisältävässä ryhmässä tai kontrolliryhmässä.
Nämä ilmeisen ristiriitaiset tulokset kuvaavat ALS:n monimutkaista luonnetta. Taudin ennusteessa on tosin joitain yhteisiä piirteitä: suurempi kalorien saanti näyttää parantavan selviytymistä ALS-potilailla, vaikka kolesterolin ja parempien olosuhteiden välillä ei ole löydetty täsmällisiä suhteita.
Insuliini lisää ndogeenisen kolesterolin tuotantoa, mikä vähentää eksogeenistä kolesterolia [4]. Siten runsaasti hiilihydraatteja sisältävä ruokavalio voi olla hyödyllinen kolesterolin tuotannon lisäämiseksi. Korkean energiapitoisuuden hiilihydraattipainotteisen ruokavalion positiivisia vaikutuksia voitaisiin soveltaa vuorotellen korkean rasvapitoisuuden (runsaasti voita [56]) ketogeenisen ruokavalion kanssa joissakin ALS-tyypeissä (SOD1), mutta ei RNA-prosessointihäiriöihin liittyvissä. (TDP43, FUS ja C9orf72).
Lisää kontrolloituja satunnaistettuja tutkimuksia tarvitaan ravitsemuksen ja ketogeenisen ruokavalion täsmällisempien menetelmien tutkimiseksi ALS-terapiana.
5. Mitokondrioiden häiriöt ja ketogeeninen ruokavalio
Edellisessä osassa viittasimme mitokondrioiden rooliin neurologisessa sairaudessa, kuten ALS. On yhä enemmän todisteita siitä, että ketogeeninen ruokavalio voi parantaa mitokondrioiden toimintaa ja stimuloida mitokondriogeneesiä [57–60].
Kuten Wallace ja hänen kollegansa ovat todenneet,
”Ironista kyllä, yksi vanhimmista terapeuttisista lähestymistavoista – paasto ja ketogeeninen ruokavalio – on edelleen lupaavin hoito mitokondrioiden häiriöissä” [61].
Itse asiassa, vaikka ketogeeninen ruokavalio on terapeuttinen työkalu, joka tunnetaan 20-luvulta lähtien, sen vaikutukset mitokondrioihin ovat suhteellisen tuore tutkimushavainto.
Jotkut mitokondrioiden häiriöt voivat aiheuttaa kohtauksia, joilla on erilaisia epileptisia fenotyyppejä [62]. Ketogeenisen ruokavalion vaikutuksista mitokondriopatioihin on joitain rohkaisevia tietoja.
Kang et al. [63] osoitti, että ketogeeninen ruokavalio voisi olla turvallinen ja tehokas hoito, joka vähentää kohtauksia lapsilla, joilla on vaikeasti hoidettava epilepsia ja erilaisia hengityskompleksihäiriöitä (kompleksi I, II, IV tai yhdistetty).
Ahola-Erkkilä ym. [64] ovat hoitaneet ketogeenisellä ruokavaliolla myöhään alkavan mitokondrioiden myopatian hiirimallia. Mitokondrioiden myopatian tiedetään aiheuttavan ihmisillä autosomaalisesti hallitsevaa progressiivista oftalmoplegiaa, lisäävän lihasheikkoutta ja mitokondrioiden mtDNA-vaurioita sekä sytokromi c-oksidaasi-negatiivisia lihaskuiduituja.
Ketogeeninen ruokavalio vähensi sytokromi c-oksidaasi-negatiivisten lihaskuitujen määrää ja esti mitokondrioiden ultrastruktuuristen poikkeavuuksien muodostumisen lihaksessa. Ruokavalio paransi suurimman osan metabolisista ja lipidoomisista poikkeavuuksista, mutta ei vaikuttamalla mtDNA:han, vaan lisäämällä mitokondrioiden biogeneesiä.
Kolikolla on kaksi puolta
Vaikka ketogeeninen ruokavalio voi olla terapeuttinen työkalu monissa mitokondriopohjaisissa sairauksissa, se on kontraindikatorinen (vasta-aiheinen; kontraindikatorinen menetelmä estää jonkin hoidon tai tutkimuksen soveltamisen, koska voi laukaista toisen häiriön tai sairauden).
Rasva-aineenvaihduntasairaudesta kärsivät potilaat saattavat kokea ketogeenisellä ruokavaliolla vakavan katabolisen kriisin. Synnynnäiset virheet lipidien aineenvaihduntaan liittyvissä entsyymeissä: mitokondrioiden kalvon pitkäketjuisten rasvahappojen kuljetusmekanismista beeta-hapetukseen ja Krebsin sykliin voivat olla kohtalokkaita paaston tai ketogeenisen ruokavalion aikana. Potilaan karnitiinipuutos, karnitiinipalmitoyylitransferaasin (CPT) I tai II puutos, karnitiinitransokaasipuutos, beta-oksidaation virheet tai pyruvaattikarboksylaasipuutos tulisi seuloa ennen ketogeenisen ruokavalio-terapian aloittamista. Ketogeeninen ruokavalio voi pahentaa myös akuuttia ajoittaista porfyriaa sairastavien potilaiden tilaa [65].

6. Alzheimerin tauti (AD) ja ketogeeninen ruokavalio
Alzheimerin tauti (AD) on yleisin neurodegeneratiivinen sairaus ja ikääntyneen väestön johtava dementian syy. AD-oireet ovat yleensä kognitiivisia häiriöitä, joihin liittyy progressiivisia muistivajeita ja persoonallisuuden muutoksia.
Alzheimer-potilaiden kognitiivisen taantumisen syyt johtuvat etenevästä synaptisesta toimintahäiriöstä ja sen aiheuttamasta hermosolujen surkastumisesta. Hermosolujen atrofiaa tapahtuu monilla aivojen alueilla: lähinnä neokorteksissa, limbisessä järjestelmässä ja aivokuoren alapuolisilla alueilla [66].
Hippokampus on ketogeenisen ruokavalion spesifinen kohde
McDaniel et al. osoitti, että kainihappo-indusoidussa status epilepticus -rottamallissa ketogeeninen ruokavalio esti mTOR-reitin signalointia aivoissa estäen hippokampuksen myöhäisen mTOR-aktivaation kainihappo-indusoidun status epilepticuksen jälkeen [59].
| Kaiinihappo on voimakas neurotoksinen aminohapon agonisti , joka toimii aktivoimalla glutamaatti-reseptoreita. Glutamaatti on pääasiallinen eksitatorinen välittäjäaine keskushermostossa. Glutamaattireseptoreihin on neljä pääluokkaa:
– NMDA-reseptorit,
– AMPA-reseptorit,
– kainaattireseptorit ja
– metabotrooppiset glutamaattireseptorit
Kainiinihappo on kainaattireseptoreiden agonisti. Kainaattireseptorit kontrolloivat todennäköisesti natriumkanavaa, joka tuottaa viritystä aiheuttavia postsynaptisia potentiaaleja (EPSP), kun glutamaatti sitoutuu. |
Hippokampuksen neuroneilla on kriittinen rooli oppimisessa ja muistin toiminnassa. Ne ovat erityisen alttiita Alzheimerin taudin aiheuttamille häiriöille ja rappeutumiselle.
AD on luokiteltu kahteen päämuotoon: familiaalinen AD (FAD) ja satunnainen AD (SAD) tai myöhään alkava ikääntymiseen liittyvä AD (LOAD); jälkimmäinen on dementian johtava syy, joka selittää yli puolet kaikista tapauksista. Melkein kaikki FAD-tapaukset johtuvat mutaatiosta kolmessa geenissä (amyloidin esiasteproteiini APP, preseniliini 1 PSEN1 ja preseniliini 2 PSEN2 [67]),
SAD:n tarkkaa etiologiaa ei ymmärretä vielä täysin. Tiedetään, että ikä on suurin riskitekijä. Alzheimerin taudin riski kasvaa eksponentiaalisesti iän myötä 65-vuotiailla tai sitä vanhemmilla ihmisillä [68].
Ikääntymisen tiedetään vaikuttavan yhdessä muiden tekijöiden kanssa. Näitä ovat:
(1) apolipoproteiini E:n (Apo E) alleelivaihtelut
(2) anatomisten reittien rappeutuminen,
(3) mitokondrioiden toimintahäiriöt,
(4) vaurioitunut veri-aivoeste,
(5) immuunijärjestelmän toimintahäiriöt,
(6) tartuntataudit ja muut ympäristötekijät, kuten altistuminen alumiinille,
(7) toistuvaT pään vammat ja
(8) aliravitsemus [69]
Kuten monissa muissakin kroonisissa sairauksissa, myös Alzheimerin taudissa hoidot voidaan jakaa kahteen luokkaan:
(A) oireenmukaiset hoidot (jotka tarjoavat tilapäistä oireiden lieventämistä muuttamatta taudin etenemistä) ja
(B) hoidot, jotka voivat mahdollisesti muuttaa taudin patogeneesiä (hidastaa tai pysäyttää taudin etenemiseen liittyviä neurologisia vaurioita)
Huolimatta joistakin FDA:n hyväksymistä lääkkeistä, kuten asetyylikoliiniesteraasin estäjistä ja memantiinista (glutamaattiantagonisti, jota käytetään käyttäytymisoireiden lieventämiseen taudin kohtalaisessa vaiheessa), tällä hetkellä ei ole olemassa tehokasta hoitoa AD:n estämiseksi, hidastamiseksi tai parantamiseksi. Suurin osa hyväksytyistä lääkkeistä tarjoaa vain kohtalaisen oireellisen vaikutuksen [70, 71].
Muiden sairauksien osalta tehokkaiden hoitojen kehittämistä vaikeuttaa AD-etiologian puutteellinen tuntemus [71] siitäkin huolimatta, että ”amyloidikaskadin” hypoteesia on tutkittu laajasti. Tämä patogeneettinen hypoteesi perustuu β-amyloidin (Aβ) neurotoksisiin ominaisuuksiin ja sen soluihin kumuloitumiseen liittyvään neurotoksisten tapahtumien kaskadin käynnistämiseen. Neurodegeneratiiviseen prosessiin lukeutuvat tunnettujen neurofibrillaaristen vyyhtien (NFT) muodostumisen lisäksi myös krooniset tulehdusreaktiot, oksidatiivisen stressin lisääntyminen ja lopuksi mitokondrioiden toimintahäiriö [71].
Alzheimerin taudin kaksi päätyyppiä johtuvat erillisistä proteiineista: tau neurofibrillaaristen vyyhtien aiheuttajana on tau-proteiini ja amyloidiplakkien tapauksessa aiheuttajana on amyloidi-β-proteiini.
Kuten edellä mainittiin, FAD:lle ja SAD:lle ei tunneta yhtenäistä etiopatogeenista mekanismia. Jälkimmäisestä on saatu havaintoja, jotka viittaavat siihen, että amyloidi-β-proteiinin ja NFT:n väheneminen toimivat yhdessä aiheuttaen mitokondrioiden toiminnan heikkenemistä ja muuttaen aivojen metabolista aktiivisuutta ikääntymisprosesseihin liittyen.
Ottaen huomioon ikääntymisprosessin ja Alzheimerin taudin välisen vahvan yhteyden ja ketogeenisen ruokavalion positiiviset vaikutukset ikääntyvissä aivoissa [72], sekä Alzheimerin taudin monitekijäisen luonteen (mitokondrioiden ja aineenvaihdunnan toimintahäiriöt), on vakuuttavaa näyttöä hypoteesille, jonka mukaan ketogeenisen ruokavalion noudattaminen AD-terapiana tuottaa myönteisiä fysiologisia, metabolisia ja kognitiivisa hoitovasteita potilailla [73, 74].
Esimerkiksi in vitro -tutkimus on osoittanut, että beeta-hydroksibutyraatin (ketoaine) lisääminen suojaa hippokampuksen hermosoluja Aβ -toksisuudelta. Tämä viittaa ketogeenisen ruokavalion mahdollisiin terapeuttisiin hyötyihin Alzheimerin tautiin liittyvissä mitokondrioiden toimintahäiriöissä [75].
Toisaalta eläinkokeet ovat antaneet osin ristiriitaisia tuloksia:
Van der Auwera et al. [76] osoitti Aβ: n vähenemistä nuorten 1,5 kk KD:lla syötettyjen siirtogeenisten AD-hiirten aivoissa, kun taas ikääntyneille koirille KD:n vaikutus näytti rajoittuneen aivojen parietaaliseen lohkoon [77].
Eläinkokeissa ketoniestereitä sisältävä pitkäaikainen (8 kuukautta) ruokinta keski-ikäisillä (8,5 kuukauden ikäisillä) hiirillä paransi hiirten kognitiota ja Aβ- ja tau-patologiaa [75]. Beckett et al. [78] osoitti, että AD-hiirimalli, jossa hiiriä ruokittiin runsaasti rasvaa ja vähän hiilihydraatteja sisältävällä ketogeenisellä ruokavaliolla, johti AD-hiirten parantuneisiin motorisin toimintoihin ilman muutoksia Aβ:ssä.
Eläinkokeiden keskenään ristiriitaiset tulokset voivat johtua eläinten iästä: hiiret ovat useimmiten nuoria tai keski-ikäisiä, mutta aineenvaihdunnan muutoksia esiintyy toistuvasti pääasiassa vanhuksilla.
Alzheimerin tauti liittyy myös metaboliseen dysregulaatioon ja insuliiniresistenssiin [79]. Monet tutkijat ovat osoittaneet, että ketogeeninen ruokavalio voi merkittävästi parantaa glukoosin homeostaasia vähentämällä aineenvaihdunnan häiriöitä ja insuliiniresistenssiä [80–82].
AD:ssä on toinen patofysiologinen mekanismi, joka johtuu muuttuneesta mitokondrioiden toiminnasta ja glukoosimetaboliasta: edistyneiden glykaation lopputuotteiden (AGE) kertyminen [83].
Huolimatta siitä, että AGE:n kertyminen soluihin ja kudoksiin on normaali ikääntymisen ominaisuus, tämä prosessi kiihtyy Alzheimerin taudissa. Glykaation lopputuotteita löytyy myös amyloidiplakeista ja neurofibrillaarisista punoksista. AGE-arvojen nousu voi selittää Alzheimerin taudin monia neuropatologisia muutoksia (proteiinien silloittuminen, oksidatiivisen stressin gliaalinen induktio ja hermosolujen surkastuminen ja kuolema).
Voidaan spekuloida, että ketogeenisen ruokavalion hermosoluja suojaavat neuroprotektiiviset vaikutukset ja ketogeeniseen ruokavalioon liittyvä glykeemisen kuorman väheneminen vaikuttavat suotuisasti Alzheimerin taudissa. Toinen mielenkiintoinen hypoteesi on ketogeenisen ruokavalion arvioidut vaikutukset mitokondriogeneesiin yhdessä mitokondriokoneiston parantamisen kanssa [61, 72, 74, 84–86].
Kuten aiemmin todettiin, mitokondrioiden toimintahäiriöiden uskotaan liittyvän Alzheimerin taudin etiologiaan [72]. Iäkkäillä potilailla on havaittu selvää hermo- ja gliasolujen mitokondrioiden metabolian heikkenemistä verrattuna terveisiin nuoriin koehenkilöihin [87]. Tämä toimintahäiriö, joka liittyy mitokondrioiden glukoosi / pyruvaattihapetuksen heikentyneeseen energiantuotantoon, voi parantaa Aβ :n ja tau:n patologista kerrostumista. Heikentynyttä mitokondrioiden toimintaa voi edustaa lisääntynyt superoksidituotanto hapettumisvaurioiden vasteena, oksidatiivisen fosforylaation väheneminen ja näin ollen mitokondrioiden elektronikuljetusketjun heikkeneminen.
Muut AD: lle ominaiset glukoosimetaboliset häiriöt aivojen tietyissä osissa liittyvät mitokondrioiden toimintahäiriöihin [88]. On mielenkiintoista huomata, että aikaisempi vähentynyt glukoosin hyödyntäminen energiasubstraattina voidaan havaita FDG-PET:llä kognitioon liittyvissä kokeissa henkilöillä, joilla on tunnettu AD-historia [89].
On luultavaa, että alentunut aivojen glukoosin käyttö (hermosolujen heikentynyt glukoosinotto) edistää AD-neuropatologian kehittymistä. Aivojen glukoosimetabolian varhainen heikkeneminen voidaan havaita ennen mitattavissa olevaa kognitiivista heikkenemistä [90]. Muut todisteet tukevat tätä teoriaa, kuten alentunut pitoisuus glukoosin kuljettajia (GLUT 1 ja 2, mutta myös hermosolujen glukoosi kuljettaja GLUT 3). Alzheimerin taudissa aivoissa todettava tau-taudin hyperfosforylaatio liittyy tähän ilmiöön [91].
Aivojen aineenvaihdunnan muutos glukoosista ketogeenisen ruokavalion tuottamiin ketoaineisiin [17] on tehokas hoitomuoto tyypin I glukoosinkuljettajapuutosoireyhtymässä [92]. Ketogeeninen ruokavalio voi olla toimiva terapiavaihtoehto myös GLUT-kuljettajien puutteen aiheuttamaan hermosolujen rappeutumiseen Alzheimerin taudissa [73].
Lopuksi: vaikka suoraa näyttöä ketogeenisen ruokavalion terapeuttisista hyödyistä Alzheimerin taudin hoidossa ei ole, tämä ravitsemuksellinen lähestymistapa näyttää lupaavalta ja ansaitsee siten laajemmat kliiniset tutkimukset.

7. Parkinsonin tauti ja ketogeeninen ruokavalio
Sporadisen Parkinsonin taudin (PD) patogeneesi on yhä ratkaisematta. Tutkimukset viittaavat siihen, että ensisijainen syy on dopaminergisten* hermosolujen eksitotoksinen rappeutuminen substantia nigrassa, mikä johtaa motoriikan heikentymiseen ja lisääntyvässä määrin kognition alentumiseen sekä muihin kortikaalisen toiminnan häiriöihin.
| *Dopaminerginen: autonomisen hermoston hermosoluista dopamiinia erittävä tai sen välityksellä stimuloituva; 2. (aineista) dopamiinin tavoin vaikuttava
Dopamiinia sisältäviä neuroneja on runsaasti erityisesti keskiaivoissa substantia nigran ja tegmentumin tienoilla. Näiden aksonit haarautuvat laajalle alueelle. Aivoissa on neljä dopaminergista päärataa: mesokortikaalinen, mesolimbinen, nigrostriataalinen ja tuberoinfundibulaarinen.
Nigrostriataalisen radan tuhoutuessa ilmentyy Parkinsonin tauti. Skitsofrenian ajatellaan johtuvan mesokortikaalisen ja mesolimbisen radan dopamiinin D2-reseptorien ylistimuloitumisesta. |
Mitokondrioiden toiminnan heikentymisellä, johon liittyy substantia nigra (mustatumake), on merkittävä rooli Parkinsonin taudin kehittymisessä ja etenemisessä [94].
Kashiwaya et al. käytti heroiinianalogia 1-metyyli-4-fenyylipyridiniumia, MPP (+), joka tuottaa dopaminergisten substantia nigran eli mustatumakkeen -solujen kuoleman estämällä mitokondrioiden NADH-dehydrogenaasien monientsyymikompleksin. Tämä aiheuttaa samanlaisen oireyhtymän kuin Parkinsonin tauti viljellyissä mesenkefaalisissa neuroneissa. β-hydroksibutyraatti suojasi näitä hermosoluja MPP (+) – toksisuuden aiheuttamalta neurodegeneraatiolta [74].
Eläinmalleissa 1-metyyli-4-fenoli-1,2,5,6-tetrahydropyridiiniä (MPTP) käytetään tuottamaan ihmisen Parkinsonin taudin kaltaista oireyhtymää jäljittelevä dopaminergisten hermosolujen selektiivinen tuhoaminen mustatumakkeessa. Kuten edellä mainituissa sairauksissa, ketogeenisen ruokavalion positiiviset vaikutukset mitokondrioiden toimintaan voivat olla avaintekijä tällaisen ruokavalion terapiakäytössä, koska ketonit voivat ohittaa Parkinsonin taudin aiheuttaman kompleksin I aktiivisuuden puutteen.
Hiirikokeissa β-hydroksibutyraatin infuusio suojasi hiiriä MPTP:n aiheuttamalta dopaminergiseltä hermoston rappeutumiselta ja motorisilta häiriöiltä [49]. Lisäksi ketogeeninen ruokavalio suojasi mustatumakkeen dopaminergisiä neuroneja 6-hydroksidopamiinin neurotoksisuudelta Parkinsonin taudin rottamallissa [95].
VanItaille et al. [96] osoitti, että ihmisillä, jotka pystyvät valmistamaan ”hyperketogeenisen” ruokavalion kotona ja noudattamaan sitä 28 päivän ajan, korkea ketoaineiden pitoisuus assosioitui taudin oireiden paranemiseen yhtenäisellä Parkinsonin taudin luokitusasteikolla Unified Parkinson’s Disease Rating Scale).

8. Glykogenoosit ja ketogeeninen ruokavalio
Glykogenoosit (glykogeenin varastointisairaudet, GSD) ovat entsyymivirheistä johtuvia perinnöllisiä häiriöitä, jotka vaikuttavat glykogeenimetaboliaan ja johtavat normaalin tai epänormaalin rakenteen glykogeenin solunsisäiseen kertymiseen erilaisiin solutyyppeihin.
Klassisesti GSD numeroitiin I – VIII niiden löytämisen ja erityisen entsyymivian mukaan [97]. Viime vuosina on tunnistettu muita primaarisia glykogenooseja (GSD 0, GSD IX – XV) [98].
GSD välittyy autosomaalisena resessiivisenä, lukuun ottamatta GSD VIII, joka on X-kytketty. Toiminnallisesta näkökulmasta GSD I, III, IV, VI ja VIII / IXa voidaan ryhmitellä maksan GSD:ksi [99], koska puutteelliset entsyymit ilmentyvät enimmäkseen maksasoluissa. Kun otetaan huomioon maksan keskeinen rooli glykemian säätelyssä glykogenolyysin avulla, ei ole yllättävää, että hypoglykemia on maksan GSD:n pääasiallinen ilmenemismuoto [97, 100]. Tämä puolestaan aiheuttaa neurologisia oireita, jotka vaihtelevat kouristuksista kohtauksiin, varsinkin sairauden alkuvaiheessa. Pitkällä aikavälillä uusiutuva vaikea hypoglykemia voi aiheuttaa aivovaurioita erityisesti GSD I:ssä (von Gierken tauti, G-6-P-fosfataasin puutos), joka on yleisin maksan GSD.
GSD-hoito perustuu ruokavaliohoitoon hypoglykemian estämiseksi. Potilaita ruokitaan tärkkelyspitoisilla elintarvikkeilla ympäri vuorokauden [100, 101]. Tieteellinen perustelu ketogeenisen ruokavalion (KD) mahdolliselle käytölle johtuu varhaisesta havainnosta, jonka mukaan hypoglykemiaan liittyvät oireet paranivat iän myötä GSD:ssä [102] sekä GSD III:ssa [100].
On tunnettua, että tämä sopeutuminen tapahtuu aivoissa sekä paastotilassa että kuumeen aikana [102]. Tämä havainto tulkittiin klassisesti aivoissa tapahtuvien sopeutumisten seurauksena, joka sallii ketoaineiden lisääntyneen käytön polttoainesubstraateina glukoosin sijasta. Sama mekanismi selittää kalorirajoituksen [100] vaikutusta, joka johtaa myös mataliin verensokeritasoihin.
Mekanistinen tulkinta olisi se, että ketogeeninen ruokavalio lisää aivojen energia-aineenvaihdunnan reittien käyttöä riippumatta glykogeenin hajoamisesta. Näiden näkökohtien perusteella ketogeenistä ruokavaliota on käytetty tehokkaasti lihassolujen GSD V:n (McArdlen tauti) hoidossa [103, 104].
Ketogeenisen ruokavalion kouristuksia estävät vaikutukset ovat tunnustetaan yleisesti, vaikka mekanismeja ei ole vielä täysin selvitetty [105]. Ketogeenisen ruokavalion mahdollista käyttöä patologisissa olosuhteissa, joille on tunnusomaista krooninen hypoglykemia, tukee edelleen se, että ketogeeninen ruokavalio on standardi GLUT1-puutosoireyhtymän hoidossa [106]. Tätä voidaan pitää maksan GSD:n metabolisena fenokopiana, koska siinä verensokeria ei voida kuljettaa hermosoluihin.
Tuore tutkimus [107] arveli, että ketogeenistä ruokavaliota voidaan käyttää menestyksekkäästi vakavan GSD III:een liittyvän kardiomyopatian hoidossa. Kaiken kaikkiaan nämä havainnot saattavat kannustaa jatkotutkimuksiin ketogeenisen ruokavalion käytöstä valittujen GSD-muotojen hoidossa.

9. Loppupäätelmä
Ketogeenisen ruokavalion aiheuttamaa erikoista metabolista tilaa on tutkittu laajalti viime vuosina. Ketoaineiden pitoisuuden nousu, verensokerin aleneminen yhdessä monien tärkeiden aineenvaihduntareittien (esim. IGF-1 / AKT / mTor, AMPK / PGC1α) kanssa on osoittautunut potentiaaliseksi terapeuttiseksi aseeksi monia neurologisia ja neuromuskulaarisia sairauksia vastaan.
Nämä tutkimukset tarjoavat teoreettisen perustan ketogeenisen ruokavalion vaikutukselle useissa hermo-lihassairauksissa. Monia korkeita esteitä on kavuttava, ennen kuin näitä löydöksiä voidaan soveltaa laajasti kliiniseen käytäntöön tai kansanterveyden parantamiseen.
Ensinnäkin ketogeenisen ruokavalion tarkasta mekanismista hermo-lihassairauksien terapiana tiedetään edelleen liian vähän, ja toiseksi tällaisen ruokavalion pitkäaikaisia vaikutuksia tulisi tutkia näillä potilailla, huolimatta siitä, että meillä on vain alustavia todisteita ja todisteita, jotka perustuvat lähinnä eläinmalleihin. Saatavilla olevat tiedot osoittavat, että KD:n aineenvaihduntamekanismi joillakin neurologisilla ja hermo-lihassairauksilla voisi olla seuraava:
(1) Tehokas energialähde tietyntyyppisten hermostoa rappeuttavien sairauksien hoidolle, joille on tunnusomaista aivojen fokaalinen hypometabolia. Tällaisia ovat esimerkiksi Parkinsonin ja Alzheimerin taudit. Neuronaaliset solut pystyvät metabolisoimaan ketoaineita glukoosipuutoksen aikana.
(2) Ketonit voivat lisätä ATP-hydrolyysin vaikutusta ja korvata asetyyli-CoA:lla Alzheimerin taudille ominaisen asetyylikoliinin vähenemisen. Glukoosimetaboliaan verrattuna ketonit tuottavat alhaisempia oksidatiivisen stressin tasoja aivoissa yhdessä suuremman soluenergiantuoton ja antioksidanttikapasiteetin kanssa. Lisäksi ketoosi voi lisätä glutationiperoksidaasia hippokampussoluissa ja yleensä vähentää mitokondrioiden ROS-tuotantoa.
(3) Lisää mitokondrioiden biogeneesireittejä (AMPK:n ja PGC1a-reitin aktivoinnin kautta). Mitokondrioiden reittien parantaminen voi auttaa parantamaan aivojen ja hermosolujen aineenvaihduntaa.
(4) Ketoaineet ohittavat ALS:n luurankolihakseen ja selkäytimeen perustuvan mitokondrioiden kompleksin I aktiivisuuden vian. Viljellyissä hermosoluissa, joita hoidetaan farmakologisilla aineilla, jotka estävät kompleksin I, ketoaineiden lisäys palauttaa kompleksin toiminnan.
(5) Vähentää sytokromi-c-oksidaasi-negatiivisten lihassyiden määrää joissakin mitokondrioiden myopatioissa ja estää mitokondrioiden ultrastruktuuristen poikkeavuuksien muodostumisen.
Lopuksi uskomme, että ketogeenistä ruokavaliota tulisi tutkia syvällisemmin sen rohkaisevan potentiaalisen terapiavaikutuksen vuoksi monien hermo-lihas- ja neurodegeneratiivisten sairauksien hoidossa.
Tutkimuskatsauksen irjoittajat toteavat, ettei heillä ole omia lehmiä ojassa tai mitään taloudellisia intressejä. Minulla ketofiilistelevänä multippelisklerootikkona on, mutta tässä toimin lähinnä editorina. Artikkelin julkaisu voi tuottaa mainostuloja joitain senttejä.
Ketogeeninen ruokavalio ei paranna syntyneitä neurologisia ja neuromotorisia vaurioita, mutta on perusteltua uskoa, että se ainakin hidastaa, ellei jopa ehkäise, uusien vaurioiden syntyä. KD ei lupaa ihmettä, mutta se lupaa parempaa kuin mitä tähän asti on ollut tarjolla. Tieto lisääntyy koko ajan. Ketogeenisen ruokavalion stimuloima autofagosytoosi yhdessä neurogeneesin ja neuroplastisuuden kanssa voi ehkä korjata joitain syntyneitä vaurioita pitkällä aikavälillä. Näín toivon.
Viitteet
- N. N. Danial, A. L. Hartman, C. E. Stafstrom, and L. L. Thio, “How does the ketogenic diet work? Four potential mechanisms,” Journal of Child Neurology, vol. 28, no. 8, pp. 1027–1033, 2013. View at: Publisher Site | Google Scholar
- E. Kossoff, “The fat is in the fire: ketogenic diet for refractory status epilepticus,” Epilepsy Currents, vol. 11, no. 3, pp. 88–89, 2011. View at: Publisher Site | Google Scholar
- R. G. Levy, P. N. Cooper, and P. Giri, “Ketogenic diet and other dietary treatments for epilepsy,” Cochrane Database of Systematic Reviews, vol. 3, 2012. View at: Google Scholar
- A. Paoli, “Ketogenic diet for obesity: friend or foe?” International Journal of Environmental Research and Public Health, vol. 11, pp. 2092–2107, 2014. View at: Google Scholar
- J. C. Mavropoulos, W. S. Yancy, J. Hepburn, and E. C. Westman, “The effects of a low-carbohydrate, ketogenic diet on the polycystic ovary syndrome: a pilot study,” Nutrition and Metabolism, vol. 2, article 35, 2005. View at: Publisher Site | Google Scholar
- R. J. Klement and U. Kämmerer, “Is there a role for carbohydrate restriction in the treatment and prevention of cancer?” Nutrition & Metabolism, vol. 8, article 75, 2011. View at: Publisher Site | Google Scholar
- T. N. Seyfried, J. Marsh, L. M. Shelton, L. C. Huysentruyt, and P. Mukherjee, “Is the restricted ketogenic diet a viable alternative to the standard of care for managing malignant brain cancer?” Epilepsy Research, vol. 100, no. 3, pp. 310–326, 2012. View at: Publisher Site | Google Scholar
- A. Accurso, R. K. Bernstein, A. Dahlqvist et al., “Dietary carbohydrate restriction in type 2 diabetes mellitus and metabolic syndrome: time for a critical appraisal,” Nutrition and Metabolism, vol. 5, no. 1, article 9, 2008. View at: Publisher Site | Google Scholar
- A. Paoli, A. Rubini, J. S. Volek, and K. A. Grimaldi, “Beyond weight loss: a review of the therapeutic uses of very-low-carbohydrate (ketogenic) diets,” European Journal of Clinical Nutrition, vol. 67, no. 8, pp. 789–796, 2013. View at: Publisher Site | Google Scholar
- A. Paoli, K. Grimaldi, L. Toniolo, M. Canato, A. Bianco, and A. Fratter, “Nutrition and acne: therapeutic potential of ketogenic diets,” Skin Pharmacology and Physiology, vol. 25, no. 3, pp. 111–117, 2012. View at: Publisher Site | Google Scholar
- A. Paoli, M. Canato, L. Toniolo et al., “The ketogenic diet: an underappreciated therapeutic option?” La Clinica Terapeutica, vol. 162, no. 5, pp. e145–e153, 2011. View at: Google Scholar
- C. E. Stafstrom and J. M. Rho, “The ketogenic diet as a treatment paradigm for diverse neurological disorders,” Frontiers in Pharmacology, vol. 3, article 59, 2012. View at: Publisher Site | Google Scholar
- P. Felig, O. E. Owen, J. Wahren, and G. F. Cahill Jr., “Amino acid metabolism during prolonged starvation,” Journal of Clinical Investigation, vol. 48, no. 3, pp. 584–594, 1969. View at: Publisher Site | Google Scholar
- O. E. Owen, P. Felig, A. P. Morgan, J. Wahren, and G. F. Cahill Jr., “Liver and kidney metabolism during prolonged starvation,” The Journal of Clinical Investigation, vol. 48, no. 3, pp. 574–583, 1969. View at: Publisher Site | Google Scholar
- S. Jitrapakdee, A. Vidal-Puig, and J. C. Wallace, “Anaplerotic roles of pyruvate carboxylase in mammalian tissues,” Cellular and Molecular Life Sciences, vol. 63, no. 7-8, pp. 843–854, 2006. View at: Publisher Site | Google Scholar
- F. C. George, “Fuel metabolism in starvation,” Annual Review of Nutrition, vol. 26, pp. 1–22, 2006. View at: Publisher Site | Google Scholar
- O. E. Owen, A. P. Morgan, H. G. Kemp, J. M. Sullivan, M. G. Herrera, and G. F. Cahill Jr., “Brain metabolism during fasting,” Journal of Clinical Investigation, vol. 46, no. 10, pp. 1589–1595, 1967. View at: Publisher Site | Google Scholar
- T. Fukao, G. Mitchell, J. O. Sass, T. Hori, K. Orii, and Y. Aoyama, “Ketone body metabolism and its defects,” Journal of Inherited Metabolic Disease, 2014. View at: Publisher Site | Google Scholar
- T. Fukao, G. D. Lopaschuk, and G. A. Mitchell, “Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry,” Prostaglandins Leukotrienes and Essential Fatty Acids, vol. 70, no. 3, pp. 243–251, 2004. View at: Publisher Site | Google Scholar
- A. Paoli, L. Cenci, M. Fancelli et al., “Ketogenic diet and phytoextracts comparison of the efficacy of mediterranean, zone and tisanoreica diet on some health risk factors,” Agro Food Industry Hi-Tech, vol. 21, no. 4, pp. 24–29, 2010. View at: Google Scholar
- R. L. Veech, “The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism,” Prostaglandins Leukotrienes and Essential Fatty Acids, vol. 70, no. 3, pp. 309–319, 2004. View at: Publisher Site | Google Scholar
- R. L. Leino, D. Z. Gerhart, R. Duelli, B. E. Enerson, and L. R. Drewes, “Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain,” Neurochemistry International, vol. 38, no. 6, pp. 519–527, 2001. View at: Publisher Site | Google Scholar
- M. D. McCue, “Starvation physiology: reviewing the different strategies animals use to survive a common challenge,” Comparative Biochemistry and Physiology A: Molecular & Integrative Physiology, vol. 156, no. 1, pp. 1–18, 2010. View at: Publisher Site | Google Scholar
- K. Sato, Y. Kashiwaya, C. A. Keon et al., “Insulin, ketone bodies, and mitochondrial energy transduction,” The FASEB Journal, vol. 9, no. 8, pp. 651–658, 1995. View at: Google Scholar
- Y. Kashiwaya, K. Sato, N. Tsuchiya et al., “Control of glucose utilization in working perfused rat heart,” The Journal of Biological Chemistry, vol. 269, no. 41, pp. 25502–25514, 1994. View at: Google Scholar
- A. Paoli, L. Cenci, and K. A. Grimaldi, “Effect of ketogenic mediterranean diet with phytoextracts and low carbohydrates/high-protein meals on weight, cardiovascular risk factors, body composition and diet compliance in Italian council employees,” Nutrition Journal, vol. 10, no. 1, article 112, 2011. View at: Publisher Site | Google Scholar
- T. N. Seyfried and P. Mukherjee, “Targeting energy metabolism in brain cancer: review and hypothesis,” Nutrition and Metabolism, vol. 2, article 30, 2005. View at: Publisher Site | Google Scholar
- J. A. Vazquez and U. Kazi, “Lipolysis and gluconeogenesis from glycerol during weight reduction with very-low-calorie diets,” Metabolism: Clinical and Experimental, vol. 43, no. 10, pp. 1293–1299, 1994. View at: Publisher Site | Google Scholar
- M. A. B. Veldhorst, M. S. Westerterp-Plantenga, and K. R. Westerterp, “Gluconeogenesis and energy expenditure after a high-protein, carbohydrate-free diet,” The American Journal of Clinical Nutrition, vol. 90, no. 3, pp. 519–526, 2009. View at: Publisher Site | Google Scholar
- A. M. Robinson and D. H. Williamson, “Physiological roles of ketone bodies as substrates and signals in mammalian tissues,” Physiological Reviews, vol. 60, no. 1, pp. 143–187, 1980. View at: Google Scholar
- H. A. Krebs, “The regulation of the release of ketone bodies by the liver,” Advances in Enzyme Regulation, vol. 4, pp. 339–353, 1966. View at: Publisher Site | Google Scholar
- N. Amen-Ra, “Humans are evolutionarily adapted to caloric restriction resulting from ecologically dictated dietary deprivation imposed during the Plio-Pleistocene period,” Medical Hypotheses, vol. 66, no. 5, pp. 978–984, 2006. View at: Publisher Site | Google Scholar
- J. C. Newman and E. Verdin, “Ketone bodies as signaling metabolites,” Trends in Endocrinology and Metabolism, vol. 25, no. 1, pp. 42–52, 2014. View at: Publisher Site | Google Scholar
- S. Jäer, C. Handschin, J. St-Pierre, and B. M. Spiegelman, “AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α,” Proceedings of the National Academy of Sciences of the United States of America, vol. 104, no. 29, pp. 12017–12022, 2007. View at: Publisher Site | Google Scholar
- C. R. Benton, D. C. Wright, and A. Bonen, “PGC-1alpha-mediated regulation of gene expression and metabolism: implications for nutrition and exercise prescriptions,” Applied Physiology, Nutrition and Metabolism, vol. 33, no. 5, pp. 843–862, 2008. View at: Publisher Site | Google Scholar
- J. Yu and J. Auwerx, “Protein deacetylation by SIRT1: an emerging key post-translational modification in metabolic regulation,” Pharmacological Research, vol. 62, no. 1, pp. 35–41, 2010. View at: Publisher Site | Google Scholar
- D. L. Williamson, “Normalizing a hyperactive mTOR initiates muscle growth during obesity,” Aging, vol. 3, no. 2, pp. 83–84, 2011. View at: Google Scholar
- B. Draznin, C. Wang, R. Adochio, J. W. Leitner, and M.-A. Cornier, “Effect of dietary macronutrient composition on AMPK and SIRT1 expression and activity in human skeletal muscle,” Hormone and Metabolic Research, vol. 44, no. 9, pp. 650–655, 2012. View at: Publisher Site | Google Scholar
- J. C. Yoon, P. Puigserver, G. Chen et al., “Control of hepatic gluconeogenesis through the transcriptional coaotivator PGC-1,” Nature, vol. 413, no. 6852, pp. 131–138, 2001. View at: Publisher Site | Google Scholar
- N. B. Ruderman, X. J. Xu, L. Nelson et al., “AMPK and SIRT1: a long-standing partnership?” The American Journal of Physiology: Endocrinology and Metabolism, vol. 298, no. 4, pp. E751–E760, 2010. View at: Publisher Site | Google Scholar
- R. G. Miller, J. D. Mitchell, M. Lyon, and D. H. Moore, “Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND),” Cochrane Database of Systematic Reviews, no. 2, Article ID CD001447, 2002. View at: Google Scholar
- L. I. Bruijn, T. M. Miller, and D. W. Cleveland, “Unraveling the mechanisms involved in motor neuron degeneration in ALS,” Annual Review of Neuroscience, vol. 27, pp. 723–749, 2004. View at: Publisher Site | Google Scholar
- L. P. Rowland and N. A. Shneider, “Amyotrophic lateral sclerosis,” The New England Journal of Medicine, vol. 344, no. 22, pp. 1688–1700, 2001. View at: Publisher Site | Google Scholar
- M. Strong and J. Rosenfeld, “Amyotrophic lateral sclerosis: a review of current concepts,” Amyotrophic Lateral Sclerosis and Other Motor Neuron Disorders, vol. 4, no. 3, pp. 136–143, 2003. View at: Publisher Site | Google Scholar
- N. Siva, “Can ketogenic diet slow progression of ALS?” The Lancet Neurology, vol. 5, no. 6, article 476, 2006. View at: Google Scholar
- D. R. Rosen, T. Siddique, D. Patterson et al., “Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis,” Nature, vol. 362, no. 6415, pp. 59–62, 1993. View at: Publisher Site | Google Scholar
- P. Pasinelli, M. E. Belford, N. Lennon et al., “Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria,” Neuron, vol. 43, no. 1, pp. 19–30, 2004. View at: Publisher Site | Google Scholar
- S. Vielhaber, D. Kunz, K. Winkler et al., “Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis,” Brain, vol. 123, no. 7, pp. 1339–1348, 2000. View at: Publisher Site | Google Scholar
- K. Tieu, C. Perier, C. Caspersen et al., “D-β-Hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease,” Journal of Clinical Investigation, vol. 112, no. 6, pp. 892–901, 2003. View at: Publisher Site | Google Scholar
- Z. Zhao, D. J. Lange, A. Voustianiouk et al., “A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis,” BMC Neuroscience, vol. 7, article 29, 2006. View at: Publisher Site | Google Scholar
- W. Zhao, M. Varghese, P. Vempati et al., “Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease,” PLoS ONE, vol. 7, no. 11, Article ID e49191, 2012. View at: Publisher Site | Google Scholar
- E. I. Posse de Chaves, A. E. Rusinol, D. E. Vance, R. B. Campenot, and J. E. Vance, “Role of lipoproteins in the delivery of lipids to axons during axonal regeneration,” Journal of Biological Chemistry, vol. 272, no. 49, pp. 30766–30773, 1997. View at: Publisher Site | Google Scholar
- L. Dupuis, P. Corcia, A. Fergani et al., “Dyslipidemia is a protective factor in amyotrophic lateral sclerosis,” Neurology, vol. 70, no. 13, pp. 1004–1009, 2008. View at: Publisher Site | Google Scholar
- S. Paganoni, J. Deng, M. Jaffa, M. E. Cudkowicz, and A. Wills, “Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis,” Muscle and Nerve, vol. 44, no. 1, pp. 20–24, 2011. View at: Publisher Site | Google Scholar
- A. M. Wills, J. Hubbard, E. A. Macklin et al., “Hypercaloric enteral nutrition in patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled phase 2 trial,” The Lancet, vol. 383, no. 9934, pp. 2065–2072, 2014. View at: Google Scholar
- A. Fergani, H. Oudart, J. G. De Aguilar et al., “Increased peripheral lipid clearance in an animal model of amyotrophic lateral sclerosis,” Journal of Lipid Research, vol. 48, no. 7, pp. 1571–1580, 2007. View at: Publisher Site | Google Scholar
- K. J. Bough, J. Wetherington, B. Hassel et al., “Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet,” Annals of Neurology, vol. 60, no. 2, pp. 223–235, 2006. View at: Publisher Site | Google Scholar
- D. Y. Kim, J. Vallejo, and J. M. Rho, “Ketones prevent synaptic dysfunction induced by mitochondrial respiratory complex inhibitors,” Journal of Neurochemistry, vol. 114, no. 1, pp. 130–141, 2010. View at: Publisher Site | Google Scholar
- S. S. McDaniel, N. R. Rensing, L. L. Thio, K. A. Yamada, and M. Wong, “The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway,” Epilepsia, vol. 52, no. 3, pp. e7–e11, 2011. View at: Publisher Site | Google Scholar
- S. Srivastava, Y. Kashiwaya, M. T. King et al., “Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet,” FASEB Journal, vol. 26, no. 6, pp. 2351–2362, 2012. View at: Publisher Site | Google Scholar
- D. C. Wallace, W. Fan, and V. Procaccio, “Mitochondrial energetics and therapeutics,” Annual Review of Pathology, vol. 5, pp. 297–348, 2010. View at: Publisher Site | Google Scholar
- H. Kang, Y. Lee, and H. D. Kim, “Mitochondrial disease and epilepsy,” Brain and Development, vol. 35, no. 8, pp. 757–761, 2013. View at: Publisher Site | Google Scholar
- H. Kang, Y. Lee, H. D. Kim, J. S. Lee, and A. Slama, “Safe and effective use of the ketogenic diet in children with epilepsy and mitochondrial respiratory chain complex defects,” Epilepsia, vol. 48, no. 1, pp. 82–88, 2007. View at: Publisher Site | Google Scholar
- S. Ahola-Erkkilä, C. J. Carroll, K. Peltola-Mjösund et al., “Ketogenic diet slows down mitochondrial myopathy progression in mice,” Human Molecular Genetics, vol. 19, no. 10, Article ID ddq076, pp. 1974–1984, 2010. View at: Publisher Site | Google Scholar
- E. H. Kossoff, B. A. Zupec-Kania, P. E. Amark et al., “Optimal clinical management of children receiving the ketogenic diet: recommendations of the International Ketogenic Diet Study Group,” Epilepsia, vol. 50, no. 2, pp. 304–317, 2009. View at: Publisher Site | Google Scholar
- A. Serrano-Pozo, M. P. Frosch, E. Masliah, and B. T. Hyman, “Neuropathological alterations in Alzheimer disease,” Cold Spring Harbor perspectives in Medicine, vol. 1, no. 1, Article ID a006189, 2011. View at: Google Scholar
- P. G. Ridge, M. T. W. Ebbert, and J. S. K. Kauwe, “Genetics of alzheimer’s disease,” BioMed Research International, vol. 2013, Article ID 254954, 13 pages, 2013. View at: Publisher Site | Google Scholar
- J. Weuve, L. E. Hebert, P. A. Scherr, and D. A. Evans, “Deaths in the United States among persons with Alzheimer’s disease (2010–2050),” Alzheimer’s & Dementia, vol. 10, no. 2, pp. e40–e46, 2014. View at: Publisher Site | Google Scholar
- B. J. Balin and A. P. Hudson, “Etiology and pathogenesis of late-onset Alzheimer’s disease,” Current Allergy and Asthma Reports, vol. 14, article 417, 2014. View at: Publisher Site | Google Scholar
- D. M. Holtzman, E. Mandelkow, and D. J. Selkoe, “Alzheimer disease in 2020,” Cold Spring Harbor Perspectives in Medicine, vol. 2, no. 11, 2012. View at: Google Scholar
- D. Selkoe, E. Mandelkow, and D. Holtzman, “Deciphering Alzheimer disease,” Cold Spring Harbor perspectives in Medicine, vol. 2, no. 1, Article ID a011460, 2012. View at: Google Scholar
- J. Yao and R. D. Brinton, “Targeting mitochondrial bioenergetics for Alzheimer’s prevention and treatment,” Current Pharmaceutical Design, vol. 17, no. 31, pp. 3474–3479, 2011. View at: Publisher Site | Google Scholar
- S. A. Hashim and T. B. Vanitallie, “Ketone Body Therapy: from ketogenic diet to oral administration of ketone ester,” Journal of Lipid Research, 2014. View at: Publisher Site | Google Scholar
- Y. Kashiwaya, T. Takeshima, N. Mori, K. Nakashima, K. Clarke, and R. L. Veech, “D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease,” Proceedings of the National Academy of Sciences of the United States of America, vol. 97, no. 10, pp. 5440–5444, 2000. View at: Publisher Site | Google Scholar
- Y. Kashiwaya, C. Bergman, J. Lee et al., “A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease,” Neurobiology of Aging, vol. 34, no. 6, pp. 1530–1539, 2013. View at: Publisher Site | Google Scholar
- I. Van Der Auwera, S. Wera, F. Van Leuven, and S. T. Henderson, “A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease,” Nutrition and Metabolism, vol. 2, article 28, 2005. View at: Publisher Site | Google Scholar
- C. M. Studzinski, W. A. MacKay, T. L. Beckett et al., “Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-β precursor protein (APP) levels in the aged dog,” Brain Research, vol. 1226, pp. 209–217, 2008. View at: Publisher Site | Google Scholar
- T. L. Beckett, C. M. Studzinski, J. N. Keller, M. Paul Murphy, and D. M. Niedowicz, “A ketogenic diet improves motor performance but does not affect β-amyloid levels in a mouse model of Alzheimer’s disease,” Brain Research, vol. 1505, pp. 61–67, 2013. View at: Publisher Site | Google Scholar
- K. Akter, E. A. Lanza, S. A. Martin, N. Myronyuk, M. Rua, and R. B. Raffa, “Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment?” British Journal of Clinical Pharmacology, vol. 71, no. 3, pp. 365–376, 2011. View at: Publisher Site | Google Scholar
- H. M. Dashti, T. C. Mathew, M. Khadada et al., “Beneficial effects of ketogenic diet in obese diabetic subjects,” Molecular and Cellular Biochemistry, vol. 302, no. 1-2, pp. 249–256, 2007. View at: Publisher Site | Google Scholar
- A. Paoli, A. Bianco, K. A. Grimaldi, A. Lodi, and G. Bosco, “Long term successful weight loss with a combination biphasic ketogenic mediterranean diet and mediterranean diet maintenance protocol,” Nutrients, vol. 5, no. 12, pp. 5205–5217, 2013. View at: Publisher Site | Google Scholar
- E. C. Westman, W. S. Yancy Jr., J. C. Mavropoulos, M. Marquart, and J. R. McDuffie, “The effect of a low-carbohydrate, ketogenic diet versus a low-glycemic index diet on glycemic control in type 2 diabetes mellitus,” Nutrition and Metabolism, vol. 5, no. 1, article 36, 2008. View at: Publisher Site | Google Scholar
- V. Srikanth, A. Maczurek, T. Phan et al., “Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease,” Neurobiology of Aging, vol. 32, no. 5, pp. 763–777, 2011. View at: Publisher Site | Google Scholar
- M. Balietti, B. Giorgetti, G. Di Stefano et al., “A ketogenic diet increases succinic dehydrogenase (SDH) activity and recovers age-related decrease in numeric density of SDH-positive mitochondria in cerebellar Purkinje cells of late-adult rats,” Micron, vol. 41, no. 2, pp. 143–148, 2010. View at: Publisher Site | Google Scholar
- M. Balietti, P. Fattoretti, B. Giorgetti et al., “A ketogenic diet increases succinic dehydrogenase activity in aging cardiomyocytes,” Annals of the New York Academy of Sciences, vol. 1171, pp. 377–384, 2009. View at: Publisher Site | Google Scholar
- M. Maalouf, J. M. Rho, and M. P. Mattson, “The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies,” Brain Research Reviews, vol. 59, no. 2, pp. 293–315, 2009. View at: Publisher Site | Google Scholar
- F. Boumezbeur, G. F. Mason, R. A. de Graaf et al., “Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy,” Journal of Cerebral Blood Flow & Metabolism, vol. 30, no. 1, pp. 211–221, 2010. View at: Publisher Site | Google Scholar
- S. Hoyer, “Causes and consequences of disturbances of cerebral glucose metabolism in sporadic alzheimer disease: therapeutic Implications,” Advances in Experimental Medicine and Biology, vol. 541, pp. 135–152, 2003. View at: Google Scholar
- L. Mosconi, R. Mistur, R. Switalski et al., “Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease,” Neurology, vol. 72, no. 6, pp. 513–520, 2009. View at: Publisher Site | Google Scholar
- T. B. Vanitallie, “Preclinical sporadic Alzheimer’s disease: target for personalized diagnosis and preventive intervention,” Metabolism: Clinical and Experimental, vol. 62, supplement 1, pp. S30–S33, 2013. View at: Publisher Site | Google Scholar
- Y. Liu, F. Liu, K. Iqbal, I. Grundke-Iqbal, and C. Gong, “Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease,” FEBS Letters, vol. 582, no. 2, pp. 359–364, 2008. View at: Publisher Site | Google Scholar
- P. Veggiotti, F. Teutonico, E. Alfei et al., “Glucose transporter type 1 deficiency: Ketogenic diet in three patients with atypical phenotype,” Brain and Development, vol. 32, no. 5, pp. 404–408, 2010. View at: Publisher Site | Google Scholar
- M. A. Reger, S. T. Henderson, C. Hale et al., “Effects of β-hydroxybutyrate on cognition in memory-impaired adults,” Neurobiology of Aging, vol. 25, no. 3, pp. 311–314, 2004. View at: Publisher Site | Google Scholar
- A. Camilleri and N. Vassallo, “The centrality of mitochondria in the pathogenesis and treatment of Parkinson’s disease,” CNS Neuroscience & Therapeutics, vol. 20, no. 7, pp. 591–602, 2014. View at: Google Scholar
- B. Cheng, X. Yang, L. An, B. Gao, X. Liu, and S. Liu, “Ketogenic diet protects dopaminergic neurons against 6-OHDA neurotoxicity via up-regulating glutathione in a rat model of Parkinson’s disease,” Brain Research, vol. 1286, pp. 25–31, 2009. View at: Publisher Site | Google Scholar
- T. B. VanItallie, C. Nonas, A. Di Rocco, K. Boyar, K. Hyams, and S. B. Heymsfield, “Treatment of Parkinson disease with diet-induced hyperketonemia: a feasibility study,” Neurology, vol. 64, no. 4, pp. 728–730, 2005. View at: Publisher Site | Google Scholar
- Y. S. Shin, “Glycogen storage disease: clinical, biochemical, and molecular heterogeneity,” Seminars in Pediatric Neurology, vol. 13, no. 2, pp. 115–120, 2006. View at: Publisher Site | Google Scholar
- E. Gazzerro, A. L. Andreu, and C. Bruno, “Neuromuscular disorders of glycogen metabolism,” Current Neurology and Neuroscience Reports, vol. 13, article 333, 2013. View at: Publisher Site | Google Scholar
- T. Goldberg and A. E. Slonim, “Nutrition therapy for hepatic glycogen storage diseases,” Journal of the American Dietetic Association, vol. 93, no. 12, pp. 1423–1430, 1993. View at: Publisher Site | Google Scholar
- S. Heller, L. Worona, and A. Consuelo, “Nutritional therapy for glycogen storage diseases,” Journal of Pediatric Gastroenterology and Nutrition, vol. 47, pp. S15–S21, 2008. View at: Publisher Site | Google Scholar
- T. J. Triomphe, “Glycogen storage disease: a basic understanding and guide to nursing care.,” Journal of pediatric nursing, vol. 12, no. 4, pp. 238–249, 1997. View at: Publisher Site | Google Scholar
- J. B. Walter, General Pathology, 1987.
- V. Busch, K. Gempel, A. Hack et al., “Treatment of glycogenosis type V with ketogenic diet,” Annals of Neurology, vol. 58, no. 2, article 341, 2005. View at: Google Scholar
- M. Vorgerd and J. Zange, “Treatment of glycogenosys type V (McArdle disease) with creatine and ketogenic diet with clinical scores and with 31P-MRS on working leg muscle,” Acta Myologica, vol. 26, no. 1, pp. 61–63, 2007. View at: Google Scholar
- J. M. Rho and R. Sankar, “The ketogenic diet in a pill: is this possible?” Epilepsia, vol. 49, supplement 8, pp. 127–133, 2008. View at: Publisher Site | Google Scholar
- P. Veggiotti and V. De Giorgis, “Dietary treatments and new therapeutic perspective in GLUT1 deficiency syndrome,” Current Treatment Options in Neurology, vol. 16, no. 5, p. 291, 2014. View at: Publisher Site | Google Scholar
- V. Valayannopoulos, F. Bajolle, J. Arnoux et al., “Successful treatment of severe cardiomyopathy in glycogen storage disease type III with d,l-3-hydroxybutyrate, ketogenic and high-protein diet,” Pediatric Research, vol. 70, no. 6, pp. 638–641, 2011. View at: Publisher Site | Google Scholar

Antonio Paoli, Antonino Bianco, Ernesto Damiani, and Gerardo Bosco
Department of Biomedical Sciences, University of Padova, Via Marzolo 3, 35031 Padova, ItalySport and Exercise Sciences Research Unit, University of Palermo, Via Eleonora Duse 2, 90146 Palermo, Italy
Academic Editor: Giuseppe D’Antona
Received24 Apr 2014
Accepted30 May 2014
Published03 Jul 2014
Copyright © 2014 Antonio Paoli et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |