Äkkiväärän virusoppi: virologian alkeet
Virologia ei ole mediaseksikkäin tutkimuksenala, mutta virusten ja virologian ymmärtäminen on ajankohtaista. Virologeista on koronapandemian seurauksena tullut mediassa paistattelevia julkkiksia ja taviksista somessa luennoivia virologian asiantuntijoita. Tämä on johtanut epäselvyyksiin, virheellisiin mielikuviin ja mielikuvituksellisiin teorioihin. Lähes jokaisella on vahva mielipide maailmasta, koronapandemiasta ja viruksista.
Mielipiteitä saa ja pitääkin olla, mutta tokentube-akatemian lyhyt oppimäärä ei kumoa virologiaan liittyviä vuosikymmenien tutkimuksia ja tutkimusten tuloksia.
Tosiasiassa virologia ei ole helppoa. Monet koronapandemiaa ahkerasti somessa kommentoivat eivät näytä ymmärtävän eroa virusten ja bakteerien tai luontaisen ja hankinnaisen immuniteetin välillä. Osa somen virologeista uskoo immuniteetin läpäisemättömäksi jedivoimakentäksi, joka vahvistuu jokaisen infektion seurauksena. Näin ei ole. Virusinfektiot altistavat kroonistuville jälkitaudeille, keskushermoston sairauksille, autoimmuunitaudeille ja monille syöville.
Viruksiin ei kuitenkaan päde yksinkertaistava hyvä-paha-jaottelu. Monet ihmisille haitalliset virukset ovat haitallisia, koska ne ovat hypänneet ihmisiin jostain muusta lajista, kuten linnuista, lepakoista tai sioista. Suurin osa viruksista on ihmisille harmittomia ja osa mahdollisesti hyödyllisiä tai välttämättömiä.
Mitä viruksista tiedetään? Kokosin katsauksen viruksista ja virologiasta. Aloitin Äkkiväärän virusopin tutustumalla Columbian yliopiston virologian professori Vincent Racaniellon kattavan luentosarjan ensimmäiseen osaan ja Molecular Medicine:n 4. painoksen virusten lisääntymistä käsittelevään lukuun. Käsittelen tässä viruksia ja virologiaa myös yleisellä tasolla.
Aihetta sivuavista Lähetti-RNA:sta ja sitä hyödyntävistä koronarokotteista voit lukea täältä. Tämä selvittää eräitä käsitteitä ja molekyylibiologian prosesseja.
Johdanto
Viruksia on kaikkialla. Ne ympäröivät meitä silmille näkymättömänä pilvenä pölyhiukkasissa ja hengitysilman aerosoleissa. Viruksia on maaperässä, ravinnossa, ilmassa, vedessä, bakteereissa, eläimissä ja ihmisissä. Jokaisen elävän organismin genomi sisältää virusten geneettistä materiaalia. Syömme ja hengitämme miljardeja viruspartikkeleita päivittäin.
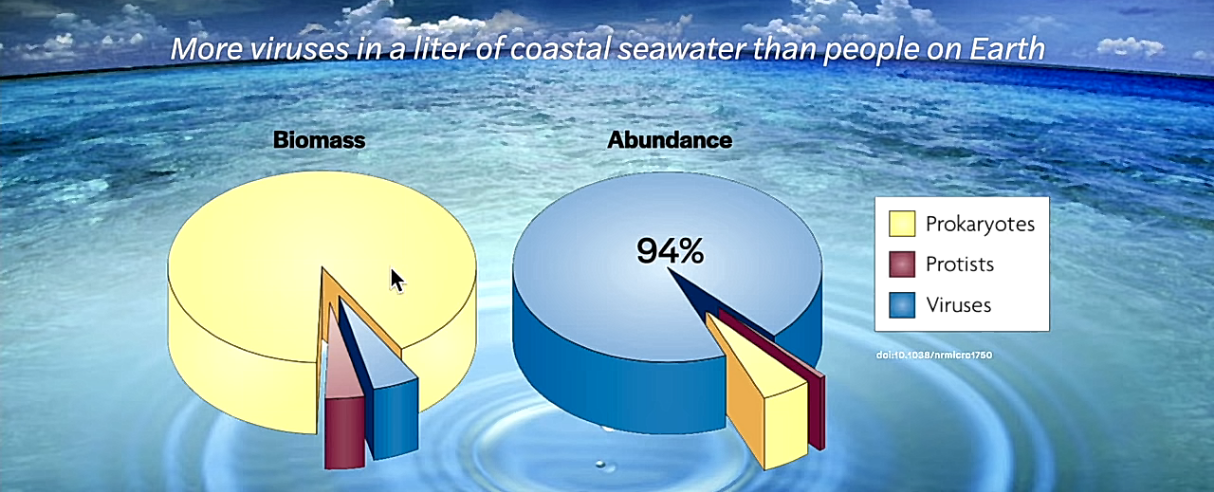
Pelkästään maapallon vesistöissä on arviolta 1030 bakteriofagia eli faagia. Ne ovat bakteereissa loisivia ja bakteereita tuhoavia viruksia. Bakteriofagi painaa vain femtogramman, eli 10-15 grammaa. Näiden bakteereissa loisivien virusten yhteenlaskettu biomassa on 1000-kertaa suurempi kuin kaikkien maailman norsujen yhteenlaskettu paino.
Kaikkien faagien jono olisi 100 miljoonaa valovuotta pitkä. Litrassa merivettä on enemmän viruksia kuin maailmassa on ihmisiä. Virusten tappamien bakteerien tuottama orgaaninen jäte vapauttaa vesistöihin ja ilmakehään niin paljon hiilidiosidia, fosforia ja rikkiä, että ne vaikuttavat ilmakehän koostumukseen ja ilmastoon.
SARS-CoV-2-pandemian vuoksi virukset nähdään helposti uhkaavina, mutta tosiasiassa suurin osa viruksista ei vaikuta ihmisiin mitenkään. Osa viruksista on todennäköisesti ihmisille hyödöllisiä ja ehkäpä jopa elintärkeitä.
Pieni osa viruksista on ihmisille haitallisia. Koronapandemia on mediassa syrjäyttänyt lähes täysin HIV-epidemiasta uutisoinnin, vaikka Afrikassa HIV:tä sairastaa 37 miljoonaa ihmistä. HIV ei häviä maailmasta luultavasti koskaan. Tehokkaat HIV:n ja AIDSin hoitoon käytettävät antiviraaliset lääkkeet näyttävät menettävän tehoaan uusien HIV-mutaatioiden kehittyessä.
Planeetallamme on arviolta 1032 yksittäistä virusta. Luku on niin käsittämättömän suuri, että se pitää määritellä ymmärrettävään muotoon: viruksia on enemmän kuin tähtiä maailmankaikkeudessa. Niitä on niin paljon, että jos maapallon kaikki virukset jaettaisiin tasan jokaiselle universumin tähdelle, kullekin tähdelle riittäisi 100 miljoonaa virusta kertoo National Geographic.
Viruksia on arviolta sata kvintiljoonaa. Vaikka ne ovat kooltaan vain muutamien kymmenien nanometrien (millimetrin miljoonasosien) mittaisia, kaikista maapallon viruksista voitaisiin muodostaa jono, joka yltäisi Linnunradalta Andromedan galaksiin asti kertoo Jyväskylän yliopiston akatemiatutkija Matti Jalasvuori.
Submikroskooppisia viruksia löytyy kaikkialta maapallolta; merivedestä ilmakehään ja maaperään. Sen lisäksi, että viruksia on käsittämättömän paljon, ne ovat aivan järjettömän pieniä. Virukset ovat niin pieniä, että niitä ei voi nähdä tavallisella mikroskoopilla.
Virukset eivät määritelmällisesti ole elollisia olentoja, koska niiltä puuttuu elämälle tunnusomaisia piirteitä, kuten aineenvaihdunta ja kyky lisääntyä itsenäisesti. Toisaalta viruksilla on myös elämään viittaavia piirteitä, kuten genomi (DNA:n tai RNA:n muodossa), joka muuttuu luonnonvalinnan vaikutuksesta.
Virusten alkuperästä ei vallitse yksimielisyyttä, mutta monet uskovat virusten kehittyneen ennen ensimmäisiä soluja. Virukset voisivat tämän hypoteesin mukaan olla elämän yksinkertaisimpia rakennuspalikoita.
Virus voi lisääntyä vain kaappaamassaan isäntäorganismin kohdesolussa. Yleensä virukset eivät tartu eri lajien välillä, mutta zoonoosit virukset voivat hypätä lajista toiseen. SARS-CoV-2 on todennäköisesti hypännyt lepakoista jonkin toisen eläimen välityksellä ihmiseen ja muuttunut ihmisten välillä tarttuvaksi viruskannaksi. Esimerkiksi eräät influenssavirukset voivat siirtyä linnuista ihmisiin tai linnuista sikoihin ja sioista ihmisiin isäntäorganismin kohdesolujen reseptorien rakenteesta riippuen.
Käytännössä eri viruslajit voivat siis käyttää lisääntymiseen elämän puun kaikkien oksien soluja isäntäsoluina. Virukset voivat levitä eläimissä, kasveissa ja jopa bakteereissa ja arkeissa. Suurin osa viruksista on ihmisille täysin harmittomia. Tämä ei johdu ihmisen sietokyvystä, vaan siitä, että virukset ovat hyvin nirsoja isäntäsolujen suhteen kertoo virologi ja tautiekologi Sara Sawyer Colorado Boulderin yliopistosta. Vain äärettömän pieni joukko viruksista voi infektoida ihmisen soluja.
Kuitenkin, kuten meneillään oleva COVID-19-pandemia selvästi osoittaa, uusia ihmisillä leviäviä viruksia löydetään vuosittain. Nykyään tunnetaan noin 200 virussukua, joiden virukset voivat infektoida ihmisen soluja ja levitä ihmisten välillä.
Mitä virologia tutkii?
Virologia tutkii submikroskooppisia, proteiinikuoren peittämiä geneettistä materiaalia sisältäviä loisorganismeja, eli viruksia [1, 2]. Virologiassa keskitytään:
- virusten rakenteeseen
- virusten luokitteluun
- virusten evoluutioon
- virusten tapaan infektoida ja hyödyntää isäntäsoluja lisääntymistä varten
- virusten vuorovaikutukseen isäntäorganismin fysiologian ja immuniteetin kanssa
- virusten aiheuttamiin sairauksiin
- virusten eristämis- ja viljelytekniikoihin
- virusten käyttöön tutkimuksessa ja terapiassa
Virologia kuuluu mikrobiologian alaan. Martinus Beijerinckin (1898) tunnistama tupakan mosaiikkitaudin bakteereista poikkeava taudinaiheuttaja tunnustetaan nyt virologian viralliseksi aluksi bakteriologiasta erillisenä tieteenalana [3, 4, 5].
Beijerinck ymmärsi, että tautia ei aiheuttanut bakteeri- tai sieni-infektio, vaan jokin muu. Beijerinck käytti sanaa ”virus” kuvaamaan salaperäistä tekijää ”contagium vivum fluidumissa” (”tarttuva elävä neste”).
Viruksen rakenne ja luokittelu
Virologian päähaara on virusten luokittelu. Virukset voidaan luokitella sen mukaan, minkä isäntäorganismin soluja ne infektoivat:
- eläinviruksiin
- kasviviruksiin
- sieniviruksiin
- bakteriofageihin (viruksiin, jotka infektoivat bakteereja, mukaan lukien monimutkaisemmat virukset). [6]
Toinen luokittelu käyttää virusten kapsidin geometrista muotoa (usein heliksi tai ikosaedri) tai viruksen rakennetta (esim. lipidivaippa – ei lipidivaippaa) [7].
Kapsidi on viruksen proteiinikuori, joka ympäröi ja suojaa viruksen genomia. Kapsidi muodostuu nukleiinihapon ympärille toistuvista, säännöllisesti järjestäytyneistä proteiinimolekyyleistä eli kapsomeereista. Se voi olla sauvamainen tai pyöreä. Genomi ja sitä ympäröivä proteiinikuori muodostavat yhdessä nukleokapsidin, jota voi eräillä viruksilla ympäröidä vielä vaippa, joka muodostuu lipidikalvosta ja siihen uponneista proteiineista.
Virukset ovat kooltaan noin 30 nanometristä noin 450 nanometriin. Suurinta osaa viruksista ei voi niiden pienen koon vuoksi tutkia perinteisillä mikroskoopeilla. Virusten muotoa ja rakennetta tutkitaan mm. elektronimikroskopialla, NMR-spektroskopialla ja röntgenkristallografialla.
Hyödyllisin ja laajimmin käytetty luokitusjärjestelmä erottaa virukset geneettisesti käyttämänsä nukleiinihapon tyypin ja viruksen replikaatiomenetelmän mukaan:
- DNA-viruksiin (jaetaan yksi- ja kaksijuosteisiin DNA-viruksiin)
- RNA-viruksiin (jaetaan positiiviseen ja negatiiviseen yksijuosteisiin RNA-viruksiin ja paljon harvinaisempiin kaksijuosteisiin RNA-viruksiin),
- käänteistranskriptoivat viruksiin (kaksijuosteiset käänteistranskriptoivat DNA-virukset ja yksijuosteiset käänteistranskriptoivat RNA-virukset, kuten retrovirukset).
Virologit tutkivat myös infektoivia subviraalisia hiukkasia, jotka ovat huomattavasti pienempiä ja yksinkertaisempia kuin virukset:
- viroidit (paljaat pyöreät RNA-molekyylit, jotka infektoivat kasveja)
- satelliitit (nukleiinihappomolekyylit kapsidin kanssa tai ilman, jotka tarvitsevat auttajavirusta infektioon ja lisääntymiseen)
- prionit (proteiinit, jotka voivat esiintyä patologisessa konformaatiossa, joka saa muut prionimolekyylit olettamaan saman konformaation) [8].
Virologian taksonit (taksoni on tieteellisessä luokittelussa käytetty termi, jolla tarkoitetaan mitä tahansa sukulaisuussuhteiden mukaan nimettyä eliöryhmää, joka on virallisesti kuvattu tieteelle taksonomisia tasoja käyttäen) eivät välttämättä ole monofyleettisiä, koska eri virusryhmien evoluutiosuhteet ovat edelleen epäselviä. Niiden alkuperästä on olemassa kolme hypoteesia:
- Virukset syntyivät elottomasta aineesta, erillään tai vielä rinnakkain solujen kanssa, ehkä viroideja muistuttavien itsereplikoituvien RNA-ribotsyymien muodossa.
- Virukset syntyivät genomin vähentymisen seurauksena aikaisemmista, toimivista soluelämän muodoista, joista tuli loisia isäntäsoluille ja jotka menettivät myöhemmin suurimman osan toiminnoistaan; esimerkkejä tällaisista pienistä loisprokaryooteista ovat Mycoplasma ja Nanoarchaea.
- Virukset syntyivät solujen liikkuvista geneettisistä elementeistä (kuten transposoneista, retrotransposoneista tai plasmideista), jotka kapseloituivat proteiinikapsideihin, saivat kyvyn ”irrota” isäntäsolusta ja infektoida muita soluja.
Erityisen kiinnostava on mimivirus, jättiläisvirus, joka leviää ameeboissa ja koodaa suurta osaa perinteisesti bakteereihin liittyvästä molekyylikoneistosta. Se voi olla yksinkertaistettu versio loisprokaryootista tai se voi olla peräisin yksinkertaisemmasta viruksesta, joka on ottanut geenejä isännästään.
Virusevoluutio tutkii virusten evoluutiota, joka tapahtuu usein yhdessä virusten isäntäorganismien evoluution kanssa. Vaikka virukset lisääntyvät ja kehittyvät, niiltä puuttuu itsenäinen aineenvaihdunta. Virukset eivät myöskään liiku itsenäisesti ja niiden lisääntyminen on riippuvainen isäntäorganismin kohdesolun proteiinikoneistosta.
Usein kiistellään siitä, ovatko virukset elossa vai eivät. Tämä on semanttinen määrittelykysymys, joka ei vaikuta virusten biologiseen todellisuuteen.
Viruksilla on eräs hyvin kiehtova ominaisuus:
1930-luvulla W.M. Stanley havaitsi, että virukset kiteytyvät tai kristalloituvat samaan tapaan kuin suola. Elollinen organismi ei kristalloitu. Tämä on eräs tapa havaita virukset. Riittävä määrä viruksia voidaan muuttaa kristallikiteiksi. Nämä kristallikiteet muuttuvat jälleen aktiivisiksi infektion aiheuttaviksi viruksiksi, kun kiteitä liotetaan nesteessä.
Virustaudit ja immuunipuolustus
Yksi tärkeimmistä syistä virusten tutkimiselle on se, että ne aiheuttavat monia tartuntatauteja, kuten flunssaa, influenssaa, raivotautia, tuhkarokkoa, monia ripulitauteja, hepatiittia, denguekuumetta, keltakuumetta, poliota, isorokkoa, AIDSia, SARSia, MERSiä ja COVID-19-tautia. [9]
Herpes simplex aiheuttaa huuliherpestä ja sukupuolielinten herpestä, ja sitä tutkitaan altistavana tekijänä Alzheimerin taudille [10].
Eräät onkogeeniset nk. onkovirukset (kasvainvirukset) altistavat syöville. Tunnetuin esimerkki on ihmisen papilloomaviruksen ja kohdunkaulan syövän välinen vahva yhteys: lähes kaikki kohdunkaulan syövät johtuvat tämän sukupuoliteitse tarttuvan viruksen spesifeistä viruskannoista. Toinen esimerkki on hepatiitti B- ja hepatiitti-C-infektioiden yhteys maksasyöpään. Myös HIV ja EBV voivat altistaa pahanlaatuisten solumuutosten kehittymiselle ja eräille syöville.
Jotkut subviraaliset hiukkaset aiheuttavat sairauksia: tarttuvat spongiformiset enkefalopatiat, joihin kuuluvat Kuru, Creutzfeldt–Jakobin tauti ja naudan spongiforminen enkefalopatia (”hullun lehmän tauti”), ovat prionien aiheuttamia [11], Hepatiitti D johtuu satelliittiviruksesta.
Virusten patogeneesi tutkii tapoja, joilla virukset aiheuttavat sairauksia. Sitä, missä määrin virus aiheuttaa tauteja määritellään virulenssilla.
Kun selkärankaisen immuunijärjestelmä kohtaa viruksen, se voi tuottaa spesifisiä vasta-aineita (immunoglobuliineja), jotka sitoutuvat virukseen ja neutraloivat sen tarttuvuuden ja merkitsevät kohtaamansa virukset tuhottavaksi.
| Esimerkiksi: mRNA-rokote vie lipidinanopartikkelissa (tai adenovirusvektorissa) SARS-CoV-2-viruksen piikkiproteiinin aminohapposekvenssin ihmisen lihassoluun. Solun sytoplasmassa lipidi hajoaa ja mRNA vapautuu tulkittavaksi. Solun ribosomit lukevat lähetti-RNA:n aminohapposekvenssin ja siirtäjä-RNA:t tuovat soluun aminohappoja, joista virukselle spesifi proteiinireseptori kootaan. Tämän translaation jälkeen mRNA hajoaa. Piikkiproteiini viedään solusta eksosytoosissa osaksi solun glykoproteiineja (MHC-kompleksiin). Immuunijärjestelmä tunnistaa MHC-kompleksiin ilmestyneen piikkiproteiinin antigeeniksi. Tämä aktivoi immuunivasteen. Immunoglobuliineja tuottavien B-solujen määrä lisääntyy ja ne alkavat valmistaa piikkiproteiinille herkistyneitä vasta-aineita. Nämä immunoglobuliinit tarttuvat piikkiproteiiniin ja estävät virusta kiinnittymästä kohdesolun ACE2-reseptoriin. Koska virukset eivät voi lisääntyä solujen ulkopuolella, vasta-aineet tukahduttavat virusten kyvyn lisääntyä ja levitä tehokkaasti. B-solut voivat tuottaa käytännössä rajattomasti erilaisia vasta-aineita. Virukseen kiinnittyneet vasta-aineet toimivat myös tunnisteina, joiden merkitsemät molekyylit makrofagit ja muut immuunijärjestelmän syöjäsolut tunnistavat vieraiksi tuhottaviksi kohteiksi. Samalla antigeenin aktivoima immuunijärjestelmä kehittää soluvälitteisen immuunivasteen, jossa sytotoksisten infektoituneita soluja tuhoavien T-solujen määrä kasvaa. Akuutin immuunivasteen loppuvaiheessa osa B- ja T-soluista erikoistuu muistisoluiksi, jotka voivat ylläpitää osittaista immuniteettia jopa vuosikymmeniä. Immunologinen muisti toimii hitaammin kuin vasta-ainevälitteinen akuutti immuunivaste, mutta se aktivoi immuunijärjestelmän tunnistettuaan aiemmin kohtaamansa taudinaiheuttajan. |
Vasta-aineiden esiintymistä veren seerumissa käytetään usein määrittämään, onko henkilö aiemmin altistunut tietylle virukselle esimerkiksi ELISA-testeillä. Rokotukset suojaavat virustaudeilta osittain indusoimalla vasta-aineiden tuotantoa. Virukselle spesifisiä monoklonaalisia vasta-aineita käytetään myös virusinfektion havaitsemiseen fluoresenssimikroskopiassa.
Selkärankaisten immuunijärjestelmän toinen mekanismi viruksia vastaan on soluvälitteinen immuunivaste, joka käyttää T-immuunisoluja (immuunijärjestelmää ohjaavia CD4-T-auttajasoluja sekä infektoituneita soluja tuhoavia CD8-T-tappajasoluja). Kehon solut esittelevät pinnallaan lyhyitä proteiineja. Jos T-solu tunnistaa sieltä epäilyttävän virusfragmentin, isäntäsolu tuhotaan ja virusspesifiset T-solut lisääntyvät. Tämä mekanismi käynnistyy infektion sairastamalla ja rokotuksilla.
RNA-interferenssi, solumekanismi, jota löytyy kasveista, eläimistä ja monista muista eukaryooteista, kehittyi todennäköisesti suojaksi haitallisia viruksia vastaan.
Vuorovaikutteisten entsyymien pitkälle kehittynyt koneisto havaitsee kaksijuosteiset RNA-molekyylit (jotka esiintyvät osana monien virusten elinkaarta) ja tuhoavat kaikki havaittujen RNA-molekyylien yksijuosteiset versiot.
Jokainen tappava virustauti on paradoksi: isännän tappamisesta ei tietenkään ole mitään hyötyä virukselle, joten miten ja miksi viruksesta tuli tappava?
Nykyään uskotaan, että useimmat virukset ovat suhteellisen hyvänlaatuisia luonnollisissa isännissään; jotkut virusinfektiot voivat jopa olla hyödyllisiä isännälle [12]. Tappavien virustautien uskotaan johtuvan hyvälaatuisen viruksen siirtymisestä lajista toiseen lajiin, joka ei ole adaptoitunut ko. virukseen (katso zoonoosi).
Esimerkiksi ihmisillä vakavaa influenssaa aiheuttavien virusten luonnolliset isäntäorganismit olivat todennäköisesti siat tai linnut. HIV:n uskotaan olevan peräisin hyvänlaatuisesta ei-ihmiskädellisen viruksesta SIV.
Vaikka (monet) virustaudit on ollut mahdollista ehkäistä rokottamalla jo pitkään, virussairauksien hoitoon tarkoitettujen antiviraalisten viruslääkkeiden kehitys on vielä lapsen kengissä.
Ensimmäinen antiviraalinen lääke oli interferoni, jota syntyy luonnollisesti, kun infektio havaitaan. Interferoni stimuloi muita immuunijärjestelmän osia.
Esimerkki tartuntamekanismista: lintuinfluenssa
Lintuinfluenssavirukset, kuten viime vuosisadalla kymmeniä miljoonia ihmisiä tappanut espanjantauti (influenssa A:n alatyyppi H1N1) voidaan tunnistaa pintaproteiinien perusteella. Pintaproteiinit muodostuvat hemagglutiniineistä (H) ja neuraminidaaseista (N). 16 tunnetusta H-tyypistä alatyyppien H5, H7 ja H9 tiedetään siirtyvän linnuista ihmisiin.
Hemagglutiniini
(HA) on influenssavirusten sekä monien muiden virusten ja bakteerien pinnoilta löydetty antigeeninen glykoproteiini. Hemagglutiniinin avulla virus kykenee sitoutumaan soluun infektoidakseen sen eli tunkeutuakseen soluun. Hemagglutiniini nimi tulee aineen kyvystä aiheuttaa punasolujen yhteenkasautumista (engl. agglutinate = liimata/liimautua yhteen).
On olemassa ainakin 16 erilaista HA-antigeenia. Nämä alatyypit merkitään H1:stä H16:een. Viimeisin alatyyppi (H16) löydettiin influenssa A-viruksista, jotka eristettiin naurulokeista Ruotsissa ja Norjassa vuonna 2005. Kolme ensimmäistä hemagglutiniinia (H1, H2 ja H3) esiintyvät ihmisten influenssaviruksissa. H1, H2 ja H3 HA-antigeenit
- tunnistavat selkärankaisten eliöiden kohdesolujen siaalihappoa sisältäviä reseptoreita, joihin kiinnittymällä virus voi tunkeutua soluun.
- mahdollistaa viruksen genomin pääsy kohdesolun sisään aiheuttamalla isäntäsolun endosomaalisen solukalvon ja viruksen ulkokalvon yhdistyminen.
Mekanismi:
HA sitoutuu vielä tunnistamattomaan glykoproteiiniin, jota on kohdesolujen pinnoilla. Tämä auttaa viruksen kiinnittymään kohdesolun pintaan. Solukalvo reagoi virukseen nielaisemalla solukalvon reseptoreihin kiinnittyneen viruksen ja osan solukalvosta endosytoosissa.
Endosytoosissa aineet siirtyvät soluun siten, että solukalvoon muodostuu pieni syvennys, joka myöhemmin kuroutuu kiinni soluun haluttava aine sisällään. Muodostunut rakkula kuroutuu irti solun sisään ja se voi luovuttaa sisältönsä jonkin toisen soluelimen jatkokäsittelyyn. Usein jatkokäsittely tapahtuu itse endosomissa.
Solun sisälle syntyy uusi kalvopäällysteinen endosomiksi kutsuttu rakkula, jossa nielaistu virus on. Solu ryhtyy sulattamaan endosomin sisältöä muuttamalla sen sisuksen happamaksi ja siirtämällä sitä kohti lysosomia. Kun endosomin pH laskee noin 6:een, viruksen HA:n alkuperäinen rakenne muuttuu epävakaaksi ja sen laskostuminen purkautuu osittain, mikä vapauttaa esille sen muutoin piilossa olevan erittäin hydrofobisen peptidiketjun osan. Tämä vapautunut niin kutsuttu fuusiopeptidi toimii kuin tarttumiskoukku kiinnittymällä ja lukittumalla endosomin seinämään. Kun HA-molekyylin loppuosa laskostuu uudelleen (alhaisemmassa happamuudessa stabiilimpaan muotoon), se vetää fuusiopeptidikoukkua aiheuttaen endosomin kalvon lähentymisen kohti viruksen omaa kalvoa. Lopulta endosomin ja viruksen kalvot yhdistyvät ja virus genomeineen vapautuu endosomista solulimaan.

Molekyylibiologia ja virusterapia
Bakteriofagit, virukset, jotka infektoivat bakteereja, voidaan suhteellisen helposti kasvattaa virusplakkeina bakteeriviljelmissä. Bakteriofagit siirtävät toisinaan geneettistä materiaalia bakteerisolusta toiseen prosessissa, joka tunnetaan nimellä transduktio [13], ja tämä horisontaalinen geeninsiirto on yksi syy, miksi ne toimivat tärkeänä tutkimustyökaluna molekyylibiologiassa.
Geneettinen koodi, ribotsyymien toiminta, ensimmäinen yhdistelmä-DNA ja varhaiset geneettiset kirjastot saatiin aikaan käyttämällä bakteriofageja. Tiettyjä viruksista peräisin olevia geneettisiä elementtejä, kuten erittäin tehokkaita promoottoreita, käytetään nykyään yleisesti molekyylibiologian tutkimuksessa.
Eläinvirusten kasvattaminen elävän isäntäeläimen ulkopuolella on vaikeampaa. Perinteisesti hedelmöitettyjä kananmunia on käytetty usein, mutta nykyään soluviljelmiä käytetään tähän tarkoitukseen yhä enemmän.
Koska joidenkin eukaryootteja infektoivien virusten on kuljetettava geneettinen materiaalinsa isäntäsolun tumaan, ne ovat houkuttelevia työkaluja uusien geenien tuomiseksi isäntään (tunnetaan nimellä transformaatio tai transfektio). Modifioituja retroviruksia käytetään usein tähän tarkoitukseen, koska ne integroivat geeninsä isännän kromosomeihin. Tätä lähestymistapaa, jossa viruksia käytetään geenivektoreina, noudatetaan geneettisten sairauksien geeniterapiassa. Ilmeinen ongelma, joka on voitettava, on transformoivan viruksen hylkääminen immuunijärjestelmän toimesta.
Faagihoito, bakteriofagien käyttö bakteerisairauksien torjuntaan, oli suosittu tutkimusaihe ennen antibioottien kehittymistä ja se on viime aikoina herättänyt uutta kiinnostusta.
Onkolyyttiset virukset ovat viruksia, jotka infektoivat syöpäsoluja. Vaikka varhaiset yritykset käyttää näitä viruksia syövän hoidossa epäonnistuivat vuosina 2005 ja 2006, niiden käytöstä on sittemmin saatu rohkaisevia alustavia tuloksia [14].
Virusten sekvensointi
Lue myös: DNA sequencing
Koska useimmat virukset ovat liian pieniä mikroskoopilla tutkittaviksi, sekvensointi on yksi virologian tärkeimmistä työkaluista viruksen tunnistamisessa ja tutkimisessa. Perinteistä Sanger-sekvensointia ja seuraavan sukupolven sekvensointia (NGS) käytetään virusten sekvensoimiseen perus- ja kliinisissä tutkimuksissa, samoin kuin uusien virusinfektioiden diagnosoinnissa, viruspatogeenien molekyyliepidemiologiassa ja lääkeresistenssitestauksessa.
GenBankissa on yli 2,3 miljoonaa ainutlaatuista virussekvenssiä. Viime aikoina NGS on ohittanut perinteisen Sangerin suosituimpana tapana luoda virusgenomeja [15].
Muut virusten käyttötarkoitukset
Äskettäin kuvattiin uusi geenimanipuloitujen virusten sovellus nanoteknologiassa; katso virusten käyttö materiaalitieteessä ja nanoteknologiassa ja neuronien kartoittamiseen käytettävät pseudorabies-sovellukset neurotieteessä.
Virologian historia
Sana virus ilmestyi vuonna 1599 ja tarkoitti alun perin ”myrkkyä” [16]. Hyvin varhainen rokotusmuoto, joka tunnetaan nimellä variolaatio, kehitettiin noin tuhat vuotta sitten Kiinassa. Se sisälsi isorokkopotilaiden rokkoeritteiden käyttämisen muiden immunisoimiseksi.
Vuonna 1717 Lady Mary Wortley Montagu tarkkaili tätä käytäntöä Istanbulissa ja yritti tehdä sen suosituksi Britanniassa, mutta kohtasi huomattavaa vastustusta. Vuonna 1796 Edward Jenner kehitti paljon turvallisemman menetelmän käyttämällä lehmärokkoa nuoren pojan menestyksekkääseen immunisointiin isorokkoa vastaan, ja tämä käytäntö otettiin laajalti käyttöön.
Seurauksena oli rokotuksia muita virustauteja vastaan, kuten Louis Pasteurin onnistunut raivotautirokotus vuonna 1886. Virusten luonne ei kuitenkaan ollut tutkijoille selvä. Vuonna 1892 venäläinen biologi Dmitri Ivanovsky käytti Chamberland-suodatinta yrittääkseen eristää tupakan mosaiikkitaudin aiheuttaneet bakteerit. Hänen kokeensa osoittivat, että infektoituneiden tupakkakasvien murskatut lehtiuutteet pysyivät tarttuvina suodatuksen jälkeen. Ivanovsky raportoi, että bakteeri saattaa tuottaa pientä tarttuvaa ainetta tai toksiinia, joka pystyy läpäisemään suodattimen [17][18][19].
Vuonna 1898 Martinus Beijerinck toisti Ivanovskyn työn, mutta meni pidemmälle ja välitti ”suodatettavan aineen” kasvilta kasveille, havaitsi toiminnan vähentyneen ja päätteli sen tarttuvaksi – replikoituvaksi isännässä – eikä siis pelkkänä myrkkynä. Hän kutsui sitä contagium vivum fluidumiksi [20].
Kysymys siitä, oliko aine ”elävä neste” vai hiukkanen, oli kuitenkin edelleen avoin. Vuonna 1903 ehdotettiin ensimmäistä kertaa, että virusten transduktio voisi aiheuttaa syöpää.
Vuonna 1908 Bang ja Ellerman osoittivat, että suodatettava virus voi välittää kanan leukemiaa. Tiedot jätettiin suurelta osin huomiotta 1930-luvulle asti, jolloin leukemiaa pidettiin syöpää aiheuttavana sairautena [21].
Vuonna 1911 Peyton Rous raportoi kanasarkooman, kiinteän kasvaimen, tarttuvan viruksen kanssa, ja siten Rousista tuli ”kasvainvirologian isä”. Virusta kutsuttiin myöhemmin Rous-sarkoomavirukseksi 1 ja sen ymmärrettiin olevan retrovirus.
Useita muita syöpää aiheuttavia retroviruksia on sittemmin kuvattu. Bakteereja (bakteriofagit) infektoivien virusten olemassaolon tunnisti ensimmäisenä Frederick Twort vuonna 1911 ja itsenäisesti Félix d’Herelle vuonna 1917. Koska bakteereja voitiin kasvattaa helposti viljelmässä, tämä johti virologian tutkimuksen räjähdysmäiseen kasvuun.
Vuoden 1918 tuhoisan espanjaninfluenssapandemian syy oli aluksi epäselvä. Vuoden 1918 lopulla ranskalaiset tutkijat osoittivat, että ”suodattimen läpäisevä virus” voi siirtää taudin ihmisiin ja eläimiin, mikä täyttää Kochin oletukset [22]. Vuonna 1926 osoitettiin, että tulirokko johtuu bakteerista, joka on saastunut tietyn bakteriofagin kautta. Vaikka kasviviruksia ja bakteriofageja voidaan kasvattaa suhteellisen helposti, eläinvirukset vaativat normaalisti elävän isäntäeläimen, mikä vaikeuttaa niiden tutkimusta valtavasti.
Vuonna 1931 osoitettiin, että influenssavirusta voidaan kasvattaa hedelmöitetyissä kananmunissa. Menetelmää käytetään edelleen rokotteiden tuottamiseen. Vuonna 1937 Max Theiler onnistui kasvattamaan keltakuumevirusta kananmunissa ja tuottamaan rokotteen heikennetystä viruskannasta; tämä rokote pelasti miljoonia ihmishenkiä ja sitä käytetään edelleen.
Max Delbrück, tärkeä bakteriofagien tutkija, kuvasi viruksen perus ”elinkaaria” vuonna 1937: ”kasvamisen” sijaan viruspartikkeli kootaan sen muodostavista osista yhdessä vaiheessa; lopulta se poistuu isäntäsolusta infektoidakseen muita soluja.
Hershey–Chasen koe vuonna 1952 osoitti, että vain DNA, ei proteiini, pääsee bakteerisoluun, kun se tarttuu bakteriofagi T2:lla. Bakteerien transduktio bakteriofagien toimesta kuvattiin ensimmäisen kerran samana vuonna.
Vuonna 1949 John F. Enders, Thomas Weller ja Frederick Robbins raportoivat polioviruksen kasvusta viljellyissä ihmisen alkiosoluissa, mikä oli ensimmäinen merkittävä esimerkki eläinten tai kananmunien ulkopuolella kasvatetusta eläinviruksesta. Tämä työ auttoi Jonas Salkia johtamaan poliorokotteen deaktivoiduista polioviruksista; Tämä rokote osoittautui tehokkaaksi vuonna 1955.
Ensimmäinen virus, joka pystyttiin kiteyttämään ja jonka rakennetta näin ollen voitiin selvittää yksityiskohtaisesti, oli tobacco mosaic virus (TMV), virus, jota Ivanovsky ja Beijerink olivat tutkineet aiemmin.
Vuonna 1935 Wendell Stanley saavutti kiteytymisensä elektronimikroskopiaa varten ja osoitti, että se pysyy aktiivisena myös kiteytymisen jälkeen. Bernal ja Fankuchen saivat selkeitä röntgendiffraktiokuvia kiteytyneestä viruksesta vuonna 1941. Tällaisten kuvien perusteella Rosalind Franklin ehdotti tupakan mosaiikkiviruksen koko rakennetta vuonna 1955. Myös vuonna 1955 Heinz Fraenkel-Conrat ja Robley Williams osoittivat että puhdistettu TMV-RNA ja sen kapsidi (kuori)proteiini voivat koota itsestään toiminnallisiksi virioneiksi, mikä viittaa siihen, että tätä kokoamismekanismia käytetään myös isäntäsolussa, kuten Delbrück ehdotti aiemmin.
Vuonna 1963 Baruch Blumberg löysi hepatiitti B -viruksen ja kehitti hepatiitti B -rokotteen. Vuoteen 1985 mennessä Harald zur Hausen oli osoittanut, että kaksi ihmisen papilloomaviruksen (HPV) kantaa aiheuttavat suurimman osan kohdunkaulan syövistä.
Vuonna 2006 julkaistiin kaksi näitä kantoja vastaan suojaavaa rokotetta. Vuosina 2006 ja 2007 raportoitiin, että pienen määrän spesifisten transkriptiotekijägeenien vieminen hiirien tai ihmisten normaaleihin ihosoluihin voi muuttaa nämä solut pluripotenteiksi kantasoluiksi, jotka tunnetaan indusoituina pluripotenteina kantasoluina. Tekniikka käyttää modifioituja retroviruksia solujen transformoimiseen; tämä on mahdollinen ongelma ihmisterapiassa, koska nämä virukset integroivat geeninsä satunnaiseen paikkaan isännän genomissa, mikä voi keskeyttää muiden geenien toiminnan ja mahdollisesti aiheuttaa syöpää [30].
Vuonna 2008 kuvattiin Sputnik-virofagi, ensimmäinen tunnettu virofagi: se käyttää auttajaviruksen koneistoa lisääntyäkseen ja estää kyseisen auttajaviruksen lisääntymisen. Sputnik lisääntyy ameebassa, jossa on mamavirus, joka on edellä mainitun mimiviruksen sukulainen ja suurin tunnettu virus tähän mennessä [31].
Endogeeninen retrovirus (ERV) on genomissa oleva viruselementti, joka on peräisin retroviruksesta, jonka genomi on liitetty jonkin organismin itulinjan genomiin ja joka siksi kopioidaan kyseisen organismin jokaisen lisääntymisen yhteydessä. On arvioitu, että noin 9 prosenttia ihmisen genomista on peräisin ERV:stä.
Vuonna 2015 osoitettiin, että ERV:n proteiinit ilmentyvät aktiivisesti 3 päivän ikäisissä ihmisalkioissa ja näyttävät vaikuttavan alkionkehitykseen ja suojaavan alkioita muiden virusten aiheuttamilta infektioilta [32].
Sen jälkeen kun Organ-on-a-chip keksittiin 2010-luvulla, tekninen lähestymistapa on löytänyt sovelluksen monien sairauksien tutkimuksessa. Lähestymistapa on otettu käyttöön myös virologiassa ja sirumalleja kehitetään. Esimerkkejä ovat Donald E. Ingber -ryhmän keksimä influenssamalli, Alireza Mashaghin ryhmän keksintö Ebola-virustautimallista ja Marcus Dornerin ryhmän keksintö virushepatiittimallista [33]. Elinsirun lähestymistapa todennäköisesti korvaa ihmisen virologian eläinmallit.
Sukelletaan syvemmälle virusten maailmaan
Dmitri Ivanovskyn vuonna 1892 julkaisemassa artikkelissa kuvattiin tupakakasveja tartuttava ei-bakteeripatogeeni. Martinus Beijerinck löysi tupakan mosaiikkiviruksen vuonna 1898.
Ympäristöstämme löytyvistä sadasta kvintiljoonasta viruksesta on yksityiskohtaisesti kuvattu ja sekvensoitu yli 9000 viruslajia.
Uskomus, että SARS-CoV-2 virusta ei ole eristetty, on rokote- ja koronakriittisten ylläpitämä harhaluulo. Koronaviruksen eristäminen edellyttää korkeimman bioturvallisuusluokan laboratorion, jollainen löytyy mm. Helsingin yliopistosta. Helsingin yliopistossa koronavirus on eristetty lukemattomia kertoja ottamalla ihmisestä nenänielutikulla näyte. Näytteelle on tehty virusviljely, jonka jälkeen virusta on kuvattu elektronimikroskoopilla.
Viruksia löytyy käytännössä kaikista maapallon ekosysteemeistä. Viruksia tutkii virologia, joka on mikrobiologian alalaji.
Tartunnan saamisen jälkeen isäntäsolu pakotetaan tuottamaan nopeasti tuhansia kopioita alkuperäisestä viruksesta. Kun viruksia ei ole infektoituneen solun sisällä tai solun tarttumisprosessissa, niitä esiintyy itsenäisten hiukkasten tai virionien muodossa, jotka koostuvat:
- geneettisestä materiaalista, ts. Pitkistä DNA:n tai RNA:n molekyyleistä, jotka koodaavat solun rakennetta. Proteiinit, joilla virus vaikuttaa
- proteiinipäällyste, kapsidi, joka ympäröi ja suojaa geneettistä materiaalia; ja joissakin tapauksissa
- lipidien ulkokuori.
Näiden viruspartikkeleiden muodot vaihtelevat yksinkertaisista kierteisistä ja ikosaedrisista muodoista monimutkaisempiin rakenteisiin. Useimmilla viruslajeilla virionit ovat liian pieniä optisella mikroskoopilla havainnointiin, koska niiden koko on vain joitain sadasosia bakteerien koosta.
Virusten alkuperä elämän evoluutiohistoriassa on epäselvä: jotkut ovat saattaneet kehittyä plasmidista – DNA-paloista, jotka voivat liikkua solujen välillä – kun taas toiset ovat saattaneet kehittyä bakteereista.
Kehityksessä virukset mahdollistavat horisontaalisen geeninsiirron, mikä lisää geneettistä monimuotoisuutta samalla tavalla kuin suvullinen lisääntyminen.
Jotkut biologit pitävät viruksia elämänmuotona, koska ne kuljettavat geneettistä materiaalia, lisääntyvät ja kehittyvät luonnollisen valinnan kautta, vaikka niiltä puuttuvatkin keskeiset elämän ominaisuudet, kuten solurakenne, joita pidetään yleensä välttämättöminä kriteereinä elämän määrittelemiseksi.
Koska viruksilla on joitain, mutta ei kaikkia elävän organismin ominaisuuksia, viruksia on kuvattu ”elämän reunalla oleviksi organismeiksi” ja itsereplikaattoreiksi.
Virukset leviävät monin tavoin. Yksi tartuntareitti kulkee tauteja kantavien organismien kautta. Tämä reitti tunnetaan vektorina: viruksia välitetään kasvista kasviin kasvimehusta ravintoa saavien hyönteisten välittämnä (esimerkiksi kirvojen välittämänä); verta imevät hyönteiset voivat puolestaan välittää viruksia eläinten välillä.
Influenssavirukset leviävät ilmassa pisara- tai aerosolitartuntoina yskimisen ja aivastuksen välityksellä. Norovirus- ja rotavirus leviävät ulosteen välityksellä esimerkiksi pilaantuneesta vedestä tai virukselle altistuneesta ravinnosta. Ihmisillä infektion tuottamiseen tarvittava tarttuva noroviruksen annos on alle 100 hiukkasta.
HIV on yksi monista viruksista, jotka tarttuvat seksuaalisen kanssakäymisen kautta tai altistumalla tartunnan saaneelle verelle.
Erilaista isäntäsolua, johon virus voi tarttua, kutsutaan sen ”isäntäalueeksi”. Tämä voi olla kapea, eli virus kykenee tartuttamaan muutamia lajeja, tai laaja, eli se kykenee tartuttamaan monia.
Eläinten virusinfektiot aiheuttavat immuunivasteen, joka yleensä eliminoi tarttuvan viruksen. Immuunivasteita voidaan tuottaa myös rokotteilla, jotka antavat keinotekoisesti hankitun immuniteetin spesifiselle virusinfektiolle. Jotkut virukset, kuten AIDSia, HPV-infektiota ja virushepatiittia aiheuttavat virukset, välttävät immuunivasteita ja johtavat kroonisiin infektioihin.
Sars-CoV-2 tarttuu herkästi ja infektio soluja tehokkaasti sille altistuneen henkilön elimistössä luoden jatkuvasti pieniä muutoksia viruksen perimäainekseen. Osa näistä muutoksista heikentää virusta, mutta suurin osa muutoksista on tarttuvuuden ja infektion voimakkuuden suhteen neutraaleja.
Isäntäorganismissa kopioituva virus tuottaa myös virusta vahventavia mutaatioita, joista tutkijat ja lääketieteellinen yhteisö ovat huolissaan. Muutaatiot voivat kiertää tai heikentää infektion tai rokotuksen antamaa immuniteettia. Muuntuneista ja tehokkaammin leviävistä viruksista voi kehittyä vallitsevia virusvariantteja, kuten koronaviruksen deltavariantin kohdalla näyttää tapahtuneen.
Virus – organismi elämän reunalla
Virus lisääntyy kiinnittymällä kohdekudoksen solujen reseptoriin ja kaappaamalla isäntäsoluja tuottamaan itsestään kopioita. Replikaatio tuottaa virheitä viruksen perimäainekseen. Nämä virheet voivat heikentää tai vahvistaa virusta.
Yleiset käsitteet Molecular Medicine (4th edition)
Virustautien patologiset vaikutukset johtuvat:
- viruksen geenituotteiden toksisesta vaikutuksesta tartunnan saaneiden solujen aineenvaihduntaan
- isännän reaktioista viruksen geenejä ilmentäviin tartunnan saaneisiin soluihin
- viruksen geneettisen materiaalin aiheuttamat solutoimintojen muutokset perimään, solusignalointiin ja proteiinisynteesiin.
Viruksen lisääntyminen
Usein akuuttien virustautien oireet ja merkit voivat olla suoraan yhteydessä viruksen aiheuttamaan solujen tuhoutumiseen. Virusten lisääntymisessä on tärkeää määritellä eräitä yleisiä käsitteitä.
Viruksen lisääntyminen edellyttää, että se voi infektoida isäntäorganismin soluja tuottamaan viruskopioita. Solujen herkkyys eri viruksille vaihtelee isäntäorganismista ja solujen kudostyypeistä riippuen.
Viruksen isäntäalue (host range) määrittää sekä ne kudossolujen tyypit että eläinlajit, joita virus voi infektoida ja joissa se voi lisääntyä. Virukset eroavat huomattavasti isäntäalueensa suhteen. Joillakin viruksilla (esim. St.Louis-enkefaliitilla) on laaja isäntäalue, kun taas toisten isäntäalue (esim. ihmisen papilloomavirukset) voi olla spesifinen joukko erilaistuneita yhden lajin soluja (esim. ihmisen keratinosyytit).
Kun ihminen, jolla on tartunnalle altistava isäntäalue, altistuu virukselle, infektoituvat solut voivat toimia viruksen sisäänkäyntiportaaleina. Näiden solujen infektio ei välttämättä riitä aiheuttamaan kliinisesti osoitettavissa olevaa tautia. Infektio voi levitä altistumisalueelta muualle elimistöön. Usein tauti kuitenkin on seurausta kohdesolujen altistumisesta virukselle.
Monissa tapauksissa (esim. hengitystieinfektioissa ja sukuelinten herpes simplex -infektioissa) viruksen kohdesolut ovat kudoksissa, jotka ensimmäisinä altistuvat virukselle. Infektion aikana virus vie soluun geneettisen materiaalinsa – RNA:n tai DNA:n -, johon liittyy usein välttämättömiä proteiineja.
Virusgenomien koot, koostumukset ja geeniorganisaatiot vaihtelevat valtavasti. Virukset näyttävät kehittyneen eri reiteillä. Kaksi tärkeää huomiota:
Ensinnäkin viruksen kyky lisääntyä ja tartunnan saaneen solun kohtalo riippuu viruksen geenituotteiden – proteiinien – synteesistä ja toiminnasta. Rakenteen ja toiminnan, geneettisen materiaalin sekvenssin ja järjestelyn sekä geenien ilmentymismekanismin välinen korrelaatio ei ole missään ilmeisempi kuin viruksissa. Niiden mekanismien monimuotoisuus, joilla virukset varmistavat proteiiniensa valmistumisen, heijastelee viruksen geneettistä rakennetta, mutta ei aina johdu siitä.
Toiseksi, vaikka virukset eroavat huomattavasti niiden sisältämien geenien lukumäärän suhteen, kaikki virukset koodaavat vähintään kolmea toimintosarjaa, jotka ilmaistaan niiden määrittelemillä proteiineilla. Virusproteiinit
- varmistavat viruksen genomien replikaation
- pakkaavat genomin viruspartikkeleiksi – virioneiksi – ja
- muuttavat infektoituneen solun rakennetta ja / tai toimintaa.
Kyky säilyä piilevänä, joka on on välttämätön ominaisuus joidenkin virusten selviytymiselle ihmispopulaatiossa, on joidenkin virusten geenituotteiden ilmaisema lisätoiminto.
Virusten käyttämä strategia näiden toimintojen varmistamiseksi vaihtelee. Muutamissa tapauksissa (papovavirukset) virusproteiinit vain auttavat isäntäentsyymejä replikoimaan virusgenomin. Useimmissa tapauksissa (esim. pikornavirukset, reovirukset, herpesvirukset) virusproteiinit replikoivat viruksen genomin, mutta jopa itsestään riippuvaisin virus käyttää tässä prosessissa ainakin joitain isäntäproteiineja.
Kaikissa tapauksissa virusproteiinit pakkaavat genomin virioneiksi, vaikka isäntäproteiinit tai polyamiinit voivat komplisoitua viruksen genomien (esim. papovavirusten) kanssa ennen viruspartikkelin biogeneesiä tai sen aikana.
Viruksen lisääntymisen vaikutukset voivat vaihdella solukuolemasta hienovaraisiin, mutta mahdollisesti erittäin merkittäviin muutoksiin solun toiminnassa ja solun pinnalla ilmentyvien antigeenien kirjoissa.
Muutama vuosi sitten ymmärryksemme virusten lisääntymisjaksoista perustui pääasiassa viljelmässä synkronisesti infektoiduissa soluissa tapahtuvien tapahtumien analyysiin; tiesimme vähän viruksista, joita ei ollut vielä kasvatettu viljellyissä soluissa.
Viime aikoina molekyylikloonaus ja viruksen geenien ilmentäminen ovat rikastuttaneet valtavasti tietämystämme viruksista, jotka lisääntyvät huonosti, jos ollenkaan (esimerkiksi ihmisen hepadnavirukset ja papilloomavirukset) viljelysoluissa.
Kaikkien virusten lisääntymisjaksoilla on useita yhteisiä piirteitä. Ensinnäkin pian tartunnan jälkeen ja jopa useita tunteja sen jälkeen vain pieniä määriä vanhempien tarttuvaa virusta voidaan havaita. Tätä väliä kutsutaan pimennysvaiheeksi; se osoittaa, että viruksen genomit ovat altistuneet niiden ilmentymiselle tarvittaville isäntäkoneille tai viruskoneille, mutta jälkeläisviruksen tuotanto ei ole vielä kasvanut havaittavalle tasolle. Pimennysvaihetta seuraa kypsymisvaihe, jossa jälkeläisvirionit kerääntyvät soluun tai solunulkoiseen ympäristöön eksponentiaalisilla nopeuksilla.
Usean tunnin (esim. pikornavirukset) tai päivien (sytomegalovirus) jälkeen lyyttisillä viruksilla infektoidut solut lopettavat kaiken metabolisen aktiivisuutensa ja menettävät rakenteellisen eheytensä.
Solut, jotka on infektoitu ei-lyyttisillä viruksilla, voivat edelleen syntetisoida viruksia. Virusten lisääntymisjakso vaihtelee 8 tunnista (picornavirukset) yli 72 tuntiin (jotkut herpesvirukset).
Viruksen saanto solua kohti vaihtelee yli 100 000 polioviruspartikkelista useisiin tuhansiin rokkovirushiukkasiin.
Virukselle herkistyneet solut
Virukselle herkistyneen solun infektio ei automaattisesti takaa, että virus lisääntyy ja että virusjälkeläisiä syntyy. Tämä on virologian tärkeimpiä käsitteellisiä kehityskulkuja, ja sitä on korostettava yksityiskohtaisesti.
Alttiiden solujen infektio voi olla tuottavaa, rajoittavaa tai keskeyttävää.
Tuottava infektio esiintyy sallivissa soluissa ja sille on ominaista tarttuvien jälkeläisten tuotanto. Keskeyttävä infektio voi esiintyä kahdesta syystä. Vaikka solu voi olla altis tartunnalle, se voi olla ei-salliva, jolloin muutama, mutta ei kaikki, virusgeenit voidaan ilmentää harvoin tunnetuista syistä. Keskeyttävä infektio voi johtua myös joko permissiivisten tai ei-permissiivisten solujen infektiosta viallisilla viruksilla, joista puuttuu virusgeenien täydellinen rakenne. Solut voivat myös olla vain ohimenevästi sallivia, ja seuraukset ovat joko se, että infektio jatkuu solussa, kunnes solusta tulee ei-salliva, tai että vain harvat soluista tuottavat viruksen jälkeläisiä.
Virologit ovat määrittäneet tämäntyyppisen infektion rajoittavaksi tai uudelleenjärjesteleväksi. Tämä luokitus ei ole triviaali; sen merkitys johtuu havainnosta, että sytolyyttiset virukset, jotka normaalisti tuhoavat sallivan solun virusreplikaatioita tuottavan infektion aikana, voivat vain vahingoittaa, mutta ei tuhota keskeyttävästi infektoituneita, sallivia tai ei-permissiivisiä soluja. Tämän vahingon seuraukset voi olla sellaisten isäntätoimintojen ilmentyminen, jotka muuttavat solun normaalista pahanlaatuiseksi. Viruksen genomien pysyvyys on yleisempi seuraus rajoittavista ja aborttisista infektioista.
Antireseptori
Kiinnittyminen muodostaa virioniproteiinin antireseptorin erityisen sitoutumisen solun pinnan reseptoriin. Klassinen esimerkki antireseptorista on influenssaviruksen hemagglutiniini.
Antireseptorit jakautuvat ihmisen ja eläimen soluja infektoivien virusten pinnoille. Monimutkaisissa viruksissa, kuten herpes simplex -viruksessa voi olla useampi kuin yksi reseptorimolekyylilaji. Antireseptoreita spesifioivien geenien mutaatiot voivat johtaa kykyyn olla vuorovaikutuksessa tiettyjen reseptorien kanssa.
Tähän mennessä tunnistetut solureseptorit ovat suurelta osin glykoproteiineja, jotka sisältävät siaalihappoa ja heparaanisulfaattia. Kiinnityminen vaatii riittävinä pitoisuuksina ioneja vähentämään sähköstaattista hylkimistä, mutta prosessi on suurelta osin lämpötilasta ja energiasta riippumaton.
Solujen altistumisherkkyyttä rajoittaa sopivien reseptorien saatavuus. Kaikki muuten herkän organismin solut eivät ekspressoi infektiolle altistavia reseptoreita. Ihmisen munuaissoluista puuttuu polioviruksen reseptoreita, mutta reseptorit ilmestyvät kun munuaissoluja viljellään soluviljelmässä.
Alttiutta ei pidä sekoittaa sallivuuteen. Vaikka solut, joilta puuttuvat reseptorit polioviruksen kiinnittymiselle ovat virukselle tunnistamattomia, ne ovat täysin sallivia, koska ne voivat tuottaa tarttuvaa virusta transfektion jälkeen esimerkiksi polioviruspartikkeleista uutetun ehjän virus-RNA:n kanssa.
Joissakin tapauksissa virusten kiinnittyminen soluihin (esim. pikornavirukset) johtavat peruuttamattomiin muutoksiin virionin rakenteessa.
Tapauksissa, joissa soluun tunkeutumista ei tapahdu, virus voi irtautua ja päätyä toiseen soluun: esimerkiksi ortomyksovirukset ja jotkut paramiksovirukset, joiden pinnalla on neuraminidaasi.
Nämä virukset voivat fuusioitua soluun reseptoreistaan katkaisemalla neuramiinihapon reseptorien polysakkaridiketjuista. Tunkeutuminen on energiasta riippuvainen vaihe. Se tapahtuu melkein välittömästi kiinnittymisen jälkeen ja sisältää yhden kolmesta mekanismista, eli
- virionin translokaation plasmamembraanin läpi
- viruspartikkelin endosytoosin, joka johtaa virionien kertymiseen sytoplasmaan vakuolien sisällä ja
- solukalvon virionin vaipan kanssa.
| Neuraminidaasit eli sialidaasit
ovat glykosidihydrolaasientsyymejä, jotka halkaisevat neuramiinihappojen glykosidisidoksia. Neuraminidaasientsyymejä on paljon ja niitä esiintyy monissa organismeissa. Yleisesti tunnetuin neuraminidaasi on virusten pinnan neuraminidaasi. Influenssavirusten pinnalta löytyviä neuraminidaaseja käytetään usein antigeenideterminantteina eli epitooppeina. Jotkut influenssavirusten neuraminidaasit antavat virukselle enemmän virulenssia kuin toiset neuraminidaasit. Virusten neuraminidaasit katalysoivat vastavalmistuneiden virionien ja isäntäsolun reseptorien terminaalisten sialihappotähteiden hydrolyysiä. Virusten neuraminidaasien aktiviteetteihin kuuluvat viruspartikkeleiden kulkeutumisen avustaminen hengitysteiden liman läpi ja virionin tuottamien jälkeläisten poistumisen avustaminen infektoituneesta solusta. On olemassa kaksi neuraminidaasipääluokkaa, jotka halkaisevat joko ekso- tai endopolysiaalihappoja:
Swiss-Prot listasi 18. lokakuuta 2006 137 eri lajeilla esiintyvää neuraminidaasityyppiä. Influenssaneuraminidaaseista tunnetaan yhdeksän alatyyppiä; kaikkia yhdeksää alatyyppiä löydetään yleisesti lintujen influenssaviruksista, mutta nisäkkäiden influenssaviruksista löydetään vain tiettyjä alatyyppejä. Ihmisistä ja sioista löydetään yleisesti vain N1 ja N2 alatyyppejä. Influenssavirusten eri alatyyppien neuraminidaaseille on ominaista serologisen ristireaktiivisuuden puuttuminen ja noin 50 prosentin aminohapposekvenssihomologia. Virusten, alkueläinten, sienten, lintujen ja nisäkkäiden lisäksi neuraminidaaseja on myös bakteereissa, joissa ne ovat tärkeitä patogeenisyyden kannalta. |
Koteloimattomat virukset tunkeutuvat soluun endosytoosissa tai fuusioitumalla. Esimerkiksi polioviruksen adsorptiossa soluun kapsidi muuttuu ja menettää eheytensä siirtyessään sytoplasmaan.
Viruksilla, jotka tunkeutuvat fuusioitumalla plasmamembraaniin (esim. herpesvirukset), vaippa pysyy plasmamembraanissa, kun taas viruksen sisäiset ainesosat valuvat sytoplasmaan. Viruksen fuusio plasmamembraaniin vaatii viruksen vaipassa olevien spesifisten virusproteiinien vuorovaikutuksen solukalvon proteiinien kanssa.
Virus-solufuusioita tapahtuu useiden virusinfektioiden aikana. Esimerkiksi HIV infektoi soluja fuusioitumalla immuunijärjestelmän solujen kalvoihin. HIV:n fuusioitumisen edellytyksenä on, että virus kykenee sitoutumaan CD4-, CCR5- ja CXCR4-reseptoreihin. Proteiineja, joiden avulla virus- tai solukalvot voivat voittaa fuusioesteet, kutsutaan fusogeeneiksi. Viruksen fuusioproteiinit ovat välttämättömiä kalvofuusion tapahtumiseksi. On näyttöä siitä, että nisäkäslajit ovat saattaneet sisällyttää nämä samat proteiinit omiin soluihinsa infektion seurauksena. Fusogeenit reagoivat ärsykkeisiin, kuten matala pH tai viruksen sitoutuminen solureseptoreihin. Ärsykkeet muuttavat konformaatiota. Konformaatiomuutos mahdollistaa fusogeenien hydrofobisten alueiden paljastamisen, jotka normaalisti olisivat sisäisesti piilossa johtuen energeettisesti epäsuotuisista vuorovaikutuksista sytosolin tai solunulkoisen nesteen kanssa. Hydrofobiset alueet tunnetaan fuusiopeptideinä tai fuusiosilmukoina, ja ne ovat vastuussa paikallisen kalvon epävakauden ja fuusion aiheuttamisesta.
Päällystäminen on yleinen termi, jota käytetään tunkeutumisen jälkeisistä tapahtumista, jotka mahdollistavat viruksen genomille ekspression. Useimpien virusten tapauksessa virioni hajoaa yksin tai solukomponenttien (entsyymien) avulla ja vain nukleiinihappo tai nukleiinihappo-proteiinikompleksi säilyy viruspartikkelista ennen virustoimintojen ilmentymistä. Adenoviruksen, herpesviruksen ja papilloomaviruksen nukleo-kapselit kulkeutuvat tuman huokosiin, joiden kautta virus-DNA vapautuu suoraan tumaan.
Ortomyksoviruksilla infektoiduissa soluissa partikkeli otetaan endosyyttiseen rakkulaan. Viruksen vaippaan upotettu ionikanava happamoittaa viruspartikkelin, muuttaa hemagglutiniinin rakennetta ja mahdollistaa viruskuoren fuusion vesikkelikalvon kanssa ja viruksen ribonukleoproteiinin (RNP) vapautumisen sytoplasmaan.
Reoviruksista poistetaan poikkeustapauksissa vain kapsidin osat, ja viruksen genomi ekspressoi kaikki toiminnot, vaikka sitä ei koskaan vapauteta täysin kapsidista. Rokkoviruksen genomi on päällystämätön kahdessa vaiheessa. Ensimmäisessä vaiheessa ulkopäällyste poistetaan isäntäsolun entsyymeillä. Virus-DNA:n vapautuminen tumasta edellyttävän tartunnan jälkeen valmistettujen viruksen geenituotteiden osallistumista.

Viruksen lisääntymisstrategiat
Virukset noudattavat solutoimintojen asettamia rajoituksia. Kehityksensä aikana virukset ovat kehittäneet useita erilaisia strategioita:
- viruksen geenien koodaamiseen ja organisointiin
- viruksen geenien ilmentämiseen (ekspressioon)
- viruksen genomien replikaatioon ja
- viruksen geenien kokoamiseen ja kypsymiseen
Ennen kuin näitä tarkastellaan yksityiskohtaisesti, on toistettava, että virusproteiinien synteesi isäntäproteiinisynteesikoneiston avulla on keskeinen tapahtuma viruksen replikaatiossa.
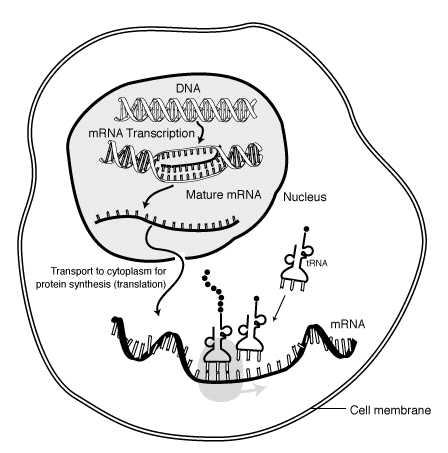
Riippumatta virusgenomin koosta, koostumuksesta ja organisaatiosta, viruksen on esitettävä kaappaamalleen solulle lähetti-RNA (mRNA), jonka solu voi tunnistaa ja kääntää (koodata proteiineiksi).
Isäntäorganismin kohdesolu asettaa viruksille kaksi rajoitusta:
Ensinnäkin solu syntetisoi oman mRNA:nsa solun tumassa transkriptoimalla DNA:n, jota seuraa transkription jälkitranskriptio, jossa mRNA luetaan ja siitä valmistetaan proteiini. Soluista puuttuu siten (a) entsyymit, jotka ovat välttämättömiä mRNA viruksen synteesille RNA-genomista tumassa tai sytoplasmassa, ja (b) entsyymit, jotka kykenevät transkriptoimaan virus-DNA:n sytoplasmassa.
Tämän rajoitteen seurauksena on, että vain virukset, joiden genomit koostuvat solun tuman saavuttavasta DNA:sta voivat hyödyntää solujen transkriptaaseja syntetisoidakseen mRNA:nsa.
Kaikkien muiden virusten on pitänyt kehittää oma transkriptaasi mRNA:n tuottamiseksi.
Toinen rajoitus on, että eukaryoottisolujen proteiinisynteesikoneisto osaa kääntää monokistronisia viestejä, sikäli kuin se ei yleensä tunnista sisäisiä aloituskohtia mRNA:issa. Tämän rajoituksen seuraukset ovat, että virukset ohjaavat erillisen mRNA:n synteesin kullekin polypeptidille (toiminnallisesti monokistroniset viestit) tai yhdelle tai useammalle mRNA:lle, jotka koodaavat ”polyproteiinia”, joka sitten pilkotaan yksittäisiksi proteiineiksi.
Harvinaisissa tapauksissa (esim. retrovirukset), määrittelemällä rakenteensa erityisellä kehyssiirrolla tai paramiksoviruksilla lisäämällä kaksi koodaamatonta nukleotidia transkriptoituun RNA:han) virusgenomin sama koodaava domeeni ohjaa kahden erillisen proteiinisarjan synteesiä.
Virukset vaihtelevat genomiensa rakenteen ja organisaation suhteen
Virusgeenit koodataan joko RNA- tai DNA-genomeihin. Nämä genomit voivat olla joko yksijuosteisia tai kaksijuosteisia. Lisäksi nämä genomit voivat olla yksiosaisia, joissa kaikki viruksen geenit sisältyvät yhteen kromosomiin, ja monipartiitteja, jossa viruksen geenit ovat jakautuneet useisiin kromosomeihin ja muodostavat yhdessä viruksen genomin.
Sekaannusten välttämiseksi ”genomiseksi” nimetään vain virioneissa esiintyvä nukleiinihappo. RNA-virusten joukossa reovirus edustaa tunnetuinta perhettä, joka sisältää kaksijuosteisen genomin, ja tämä genomi on moniosainen (koostuu 10 segmentistä tai kromosomista).
Yksisäikeisten RNA-virusten genomit ovat joko moniosaisia (pikornaviruksia, togaviruksia, paramiksoviruksia, rhabdoviruksia, koronaviruksia, retroviruksia) tai moniosaisia (ortomyksoviruksia, arenaviruksia ja bunyaviruksia).
Kaikki RNA-genomit ovat lineaarisia molekyylejä. Jotkut (esim. pikornavirukset) sisältävät kovalenttisesti kytketyn polypeptidin tai aminohapon RNA: n 5′-päässä.
Kaikki tunnetut DNA-virukset, jotka infektoivat selkärankaisia isäntiä, sisältävät moniosaisen genomin. Parvoviruksen genomeja lukuun ottamatta kaikki ovat kokonaan tai ainakin osittain kaksisäikeisiä. Yksittäiset parvovirusvirionit sisältävät lineaarista yksijuosteista DNA:ta; joissakin suvuissa (esim. adenovirus) DNA: n molemmat komplementaariset säikeet on pakattu, mutta eri viruspartikkeleihin.
Papova- ja papilloomavirusten genomit ovat suljettuja pyöreitä DNA-molekyylejä. Vaikka sekä adenovirusten että herpesvirusten genomit ovat lineaarisia kaksisäikeisiä molekyylejä, yksi juoste adenoviruksen genomin kummassakin päässä on kovalenttisesti sitoutunut proteiiniin, kun taas herpesviruksen DNA:lla on 3′-yksi nukleotidipidennys kummassakin päässä.
Rokkovirusten DNA:t ovat myös lineaarisia, mutta tässä tapauksessa kunkin juosteen 3′-pää on kovalenttisesti liitetty jatkuvan silmukan muodostavan komplementaarisen juosteen 5′-päähän.
Hepatiitti B -viruksen DNA on pyöreä kaksisäikeinen molekyyli, jossa jokaisessa juosteessa on aukko. Virukset eroavat toisistaan tavalla, jolla ne ekspressoivat geenejä ja jäljittelevät genomiaan.
Yksijuosteiset RNA-virukset
Lineaariset yksijuosteiset RNA-virukset muodostavat 3 ryhmää. Pikornavirukset ja togavirukset ovat esimerkkejä ensimmäisestä ryhmästä. Näillä genomeilla on kaksi tehtävää:
Ensimmäinen näistä toiminnoista on toimia lähetti-RNA:na. Yleensä virukset, joiden genomit voivat toimia ja toimivat lähetti-RNA:na, tunnetaan plus (+) -juosteviruksina.
Soluun tulon jälkeen pikornaviruksen RNA sitoutuu ribosomeihin ja transloituu kokonaisuudessaan. Tämän translaation tuote – polyproteiini – katkaistaan proteolyyttisillä entsyymeillä. Vaikka sekundaarisissa pilkkomisissa on mukana viruksen määrittelemiä proteaaseja, on todisteita siitä, että polyproteiini itsessään on entsymaattisesti aktiivinen trans-proteiineissa, toisin sanoen molekyyli ei pysty pilkkomaan itseään, mutta se voi pilkkoa muita polyproteiineja.
Genomisen RNA:n toinen tehtävä on toimia mallina (templaattina) komplementaarisen (-) juosteen RNA:n synteesille polymeraasin avulla, joka on johdettu polyproteiinin pilkkomisesta. (-) RNA-juoste toimii sitten vuorostaan templaattina, jotta saadaan enemmän (+) RNA-juosteita. Jälkeläisten (+) juosteet voivat sitten toimia (a) mRNA:na tai (b) templaateina, jotta saadaan enemmän (-) RNA-juosteita.
Togavirukset ja jotkut muut (+) -juosteiset RNA-virukset eroavat pikornaviruksista. Tarkemmin sanottuna vain osa genomisesta RNA:sta on saatavana translaatioksi proteiinisynteesin ensimmäisellä kierroksella. Tuloksena olevien tuotteiden todennäköinen tehtävä on transkriptoida genominen RNA, jolloin saadaan täyspitkä (-) RNA-juoste. Tämä (-) RNA-juoste toimii templaattina kahdelle (+) RNA-molekyylin kokoluokalle.
Ensimmäinen on pieni mRNA, joka kattaa genomisen RNA:n alueen, jota ei ole käännetty ensimmäisellä kierroksella. Tuloksena oleva polyproteiini pilkotaan proteiineiksi, joiden päätehtävänä on toimia virionien rakenteellisina komponentteina. (+) RNA:n toinen luokka on täysikokoinen genominen RNA, joka on pakattu virioneihin. Useita mRNA-lajeja tuotetaan koronaviruksilla, kalisiviruksilla tai hepatiitti E -viruksilla infektoiduissa soluissa.
Keskeistä (+) -juoste-virusten replikaatiossa on genomisen RNA:n kyky toimia mRNA:na infektion jälkeen. Seuraukset ovat kaksinkertaiset:
Ensinnäkin, genomin replikaatiosta vastaavat entsyymit valmistetaan infektion jälkeen, eikä virionin tarvitse tuoda niitä tartunnan saaneeseen soluun. Siksi virioneista uutettu RNA on tarttuvaa.
Toiseksi, koska kaikki (+) -juostegenomit ovat yksiosaisia, niiden kaikki geenit on kytketty yhteen kromosomiin. Sekä genomisen RNA:n että mRNA-lajien translaation alkutuotteet ovat välttämättä yksi proteiini. Pikornavirusten ja togavirusten translaatiotuotteet on katkaistava virionissa tai infektoituneessa solussa olevien yksittäisten proteiinien tuottamiseksi.
Ortomyksovirukset, paramiksovirukset, bunyavirukset, arenavirukset ja rabdo-virukset käsittävät toisen sarjan yksijuosteisia RNA-viruksia, jotka on määritelty miinus (-) juosteen viruksiksi.
(-) -juosteiset virukset on kätevä erottaa kahteen ryhmään ortomyksovirukset, bunyavirukset ja arenavirukset moniosaisista viruksista (paramykovirukset ja rabdovirukset). Tyypillisesti niiden genomisten RNA:iden on palveltava kahta templaattitoimintoa, ensimmäisessä vaiheessa mRNA:n synteesiä varten, ja toisessa komplementaaristen (+) säikeiden synteesissä, jotka toimivat templaattina viruksen jälkeläisten genomien valmistamiseksi. Koska niiden genomi on transkriptoitava mRNA:n tekemiseksi, ja solusta puuttuu sopivat entsyymit, kaikki miinusäikeiset virukset pakataan virionissa transkriptaasiin viruksen genomin ohella.
Genomin transkriptio
Viruksen genomin transkriptio on ensimmäinen tapahtuma virusten soluihin pääsyn jälkeen; prosessi tuottaa toiminnallisesti monokistroniset mRNA:t (+ säikeet), joista kukin määrittelee yhden proteiinin.
Replikaatio alkaa vasta syntetisoitujen virusproteiinien johdolla, joista valmistetaan täyspitkä (+) juoste ja se toimii templaattina (-) juosteen genomisten RNA:iden synteesille. Toistettavaksi toisin kuin + -juoste-virukset, (-) -juoste-virukset toimivat transkription mallipohjina, ensin mRNA:n synteesiin ja sitten (+)-juosteen transkriptioon, joka toimii mallina miinusjuosteelle .
Seuraukset ovat kolminkertaiset:
Ensinnäkin viruksen on tuotava infektoituneeseen soluun transkriptaasi valmistamaan mRNA:nsa. Toiseksi seuraa, että virioneista uutettu RNA ei ole tarttuvaa. Kolmanneksi tuotetut mRNA:t määrittelevät yhden polypeptidin.
RNA-silmukointisignaalien selektiivinen (mutta ei mielivaltainen) tarkkailu voi kuitenkin johtaa useisiin lähetti-RNA-molekyyleihin, joista kukin spesifioi eri proteiinin transkriptoinnin samalta genomisen RNA:n alueelta. Tämän seurauksena mRNA:na toimiva (+) transkriptio eroaa (+) juosteesta RNA:sta, joka toimii jälkeläisviruksen templaattina, vaikka molemmat syntetisoidaankin genomisella RNA:lla.
Siten moniosaisen genomin tapauksessa + -juosteisella RNA:lla, joka toimii mRNA:na, on molekyyliä suojaava huppu 5′-päässä ja poly (A)-häntä 3′-päässä, eikä se välttämättä sisällä kaikkia koodaamattomia sekvenssejä, jotka sisältyvät genomiseen RNA:han. Etuna on, että signaalit, jotka määräävät transloituneen proteiinin runsauden, upotetaan itse RNA:han. Moniosaisen (-) juosteen virusten tapauksessa mRNA koodaa vain yhtä proteiinia, mRNA:n runsaus määritetään templaatin sijainnilla genomisella RNA:lla (mitä kauempana transkription aloituskohdasta, sitä vähemmän runsasta on mRNA ja proteiinituotteen runsaus ovat suoraan yhteydessä mRNA:n runsauteen).
Kaksisuuntaiset (2 RNA-segmenttiä) arenavirukset ja jotkut kolmiosaisista (2 RNA-segmenttejä) bunyaviruksista ovat ambisenseja; ts. ne sisältävät RNA:n, jolla on sekä (+) että (-) napaisuus. Tässä tapauksessa viruksen genomi toimii aluksi, sillä sillä on (-) juosteen polaarisuus siinä mielessä, että ne transkriptoidaan tekemään (+) juosteen mRNA:ita. Nämä koodaavat proteiineja, jotka mahdollistavat komplementaarisen (+) juosteen RNA:n synteesin. Sitten (+) -juosteiset RNA:t transkriptoidaan kahden tyyppisen (-) juosteisen RNA:n muodostamiseksi. Yksi sarja toimii (-) juosteen genomisena RNA:na, joka on pakattu virioneihin. Toinen sarja edustaa ambisense-genomisen RNA: n osittaisia sekvenssejä. Vaikka määritelmän mukaan tämä RNA on (-) -juoste, koska se sisältää sekvenssejä, joilla on sama polariteetti kuin genomisella RNA:lla, se koodaa mRNA:na virusproteiineja.
Retrovirukset käsittävät kolmannen RNA-virusten ryhmän. Tyypillisesti retrovirusgenomit ovat yksiosaisia, mutta diploideja. Nämä säikeet ovat joko osittain vetysietoisia toiseen makromolekyyliin tai emäsparit pareittain vielä tuntemattomalla tavalla.
Infektion jälkeen ainoa tunnettu genomisten RNA:iden tehtävä on toimia templaattina virus-DNA:n synteesille. Siltä osin kuin eukaryoottisoluilla ei ole entsyymejä, jotka kykenisivät suorittamaan tämän tehtävän, virioni sisältää genomin lisäksi RNA-riippuvaisen DNA-polymeraasin (käänteiskopioijaentsyymin) sekä isäntänsiirto-RNA:iden seoksen, joista yksi toimii alukkeena.
Genomitranskription keskeiset vaiheet ovat:
- tRNA (eli siirtäjä-RNA)- käänteistranskriptaasikompleksin sitoutuminen genomiseen RNA: han
- genomiselle RNA:lle komplementaarisen DNA-molekyylin synteesi yhdistettynä RNA:n pilkkomiseen viruksen ribonukleaasilla (RNaasi) H, myös virioniin pakattu), spesifinen RNA:lle RNA-DNA-hybridissä, ja
- komplementaarisen DNA-juosteen synteesi ja sellaisen lineaarisen DNA-molekyylin täydentäminen, joka sisältää kokonaisuudessaan genomisen RNA:n sisältämät sekvenssit, mutta kopioimalla kahdesta pienestä sekvenssistä, yksi RNA: n 3′-päästä, joka on kopioitu DNA:n 5′-päässä, ja toinen RNA:n 5′-päästä, joka on kopioitu DNA:n 3′-päässä.
Kaksoisjuosteinen DNA siirtyy sitten tumaan, jossa virusproteiinit integroivat isäntägenomiin. Virusgeenien ilmentyminen ei välttämättä seuraa välittömästi. Kun se tapahtuu, isännän RNA-polymeraasi II transkriboi integroidun virus-DNA:n. Transkriptiotuotteet ovat genomin pituisia RNA-molekyylejä ja lyhyempiä, geeniklusterin pituisia mRNA:ita, jotka käännetään polyproteiinien tuottamiseksi.
Polyproteiinit pilkotaan sitten yksittäisten virusproteiinien tuottamiseksi. Ainakin yhden proteiinin synteesi suoritetaan ribosomaalisella kehyssiirrolla. Vain genomin pituinen transkriptio on pakattu virioneihin.
Retrovirukset vaihtelevat genomiensa monimutkaisuudessa. Kaikki retrovirusgenomit koodaavat vastuselementtejä (cis-vaikuttavat kohdat) solun transaktiotekijöille, jotka voivat olla kudosspesifisiä, ja isännän RNA-polymeraasin transkriptioaloitukselle. Monimutkaisemmat lentivirukset (retrovirusten alaryhmä) koodaavat transaktiotekijöitä, jotka säätelevät virusproteiinien runsautta ja ilmentämisjärjestystä.
Kaksijuosteiset RNA-virukset
Kaksisäikeinen, moniosainen reoviruksen genomi transkriptoidaan osittain avatussa kapsidissa virioniin pakatun polymeraasin avulla ja 10 erilaista mRNA (+ säikeet) -lajia ekstrudoidaan kapsidin paljaiden pisteiden läpi.
Lähetti-RNA-molekyyleillä on kaksi tehtävää. Ensinnäkin ne käännetään monokistronisina viesteinä virusproteiinien tuottamiseksi. Toiseksi, yksi RNA kustakin 10 lajista kokoontuu edeltäjähiukkasessa, jossa ne toimivat templaattina komplementaarisen juosteen synteesille, jolloin saadaan kaksisäikeisiä genomisegmenttejä.
DNA-viruksen genomit
DNA-virukset voidaan jakaa 4 ryhmään. Papovirus-, adenovirus- ja herpesvirusgenomit transkriptoidaan ja replikoidaan solun tumassa. Ne voivat käyttää isännän transkriptioentsyymejä mRNA:n tuottamiseen. Näiden virusten DNA:t ovat tarttuvia. Transkriptio-ohjelma koostuu vähintään kahdesta transkriptiosyklistä papoviruksille ja vähintään kolmesta herpesviruksille ja adenoviruksille. Kummassakin tapauksessa rakenteelliset tai virionipolypeptidit valmistetaan mRNA:sta, joka on muodostettu viimeisestä transkriptiosyklistä.
Rokkovirukset muodostavat toisen ryhmän. Vaikka rokkoviruksen DNA:ta on havaittu solun tumassa, transkriptio ja useimmat muut lisääntymiskierron tapahtumat näyttävät tapahtuvan sytoplasmassa. Genomin transkriptoi virusentsyymi. Alkuperäinen transkriptio tapahtuu virionin ytimessä. Monet tämän viruksen lisääntymissykliä koskevat kysymykset ovat edelleen ratkaisematta.
Parvovirukset muodostavat kolmannen ryhmän. Yksi ihmisen parvovirus, adeno-assosioitunut virus, vaatii adenoviruksia tai herpes simplex -viruksia auttajaviruksiksi lisääntyäkseen. Auttajaviruksen puuttuessa genomi näyttää integroituvan ihmisen kromosomin spesifiseen lokukseen. Muut ihmisen parvovirukset pystyvät lisääntymään ilman ”auttajaviruksen” apua.
Viruksen replikaatio sisältää yksijuosteiselle genomi-DNA:lle komplementaarisen DNA-juosteen synteesin ytimessä ja genomin transkription. Hepadnavirukset, joista esimerkkinä hepatiitti B -virus, muodostavat 4. ryhmän. Tämän viruksen DNA korjataan ja muunnetaan suljetuksi pyöreäksi molekyyliksi virioniin pakatun DNA-polymeraasin avulla, minkä jälkeen se transkriptoidaan kahdeksi RNA-molekyyliluokaksi, eli mRNA:ksi, joka ohjaa proteiinien valmistusta ja genomi-RNA:ksi, jonka käänteistranskriptaasi tuottaa genomisen DNA:n.
Virukset eroavat kokoonpanonsa, kypsymisensä ja infektoituneista soluista poistumisensa suhteen
Virukset ovat kehittäneet kaksi perustavaa laatua olevaa strategiaa niiden kokoamiseen, kypsymiseen ja vapautumiseen tartunnan saaneesta solusta.
Ensimmäinen, josta esimerkkinä vaipattomat virukset, kuten pikornavirukset, reovirukset, papovavirukset, parvovirukset ja adenovirukset, sisältää solunsisäisen kokoonpanon ja kypsymisen. Pikornavirusten tapauksessa 60 kopiota kustakin virioniproteiineista, jotka on nimetty VP0:ksi, VP1:ksi ja VP3:ksi, kerääntyy sytoplasmaan prokapsidiksi. Viruksen RNA pakataan sitten prokapsidiin ja prosessissa VP0 pilkotaan, jolloin saadaan kaksi polypeptidiä, VP2 ja VP4. Katkaisu aiheuttaa kapsidin uudelleenjärjestelyn termodynaamisesti stabiiliksi rakenteeksi, jossa RNA on suojattu nukleaasien pääsyltä.
Reovirukset kerääntyvät myös solun sytoplasmaan. Sitä vastoin adenovirukset, papovavirukset ja parvovirukset kokoontuvat tumaan. Yleensä kaikki virukset, jotka kerääntyvät ja saavat tarttuvuuden solun sisällä, ovat suurelta osin riippuvaisia tartunnan saaneen solun hajoamisesta, jolloin ne vapautuvat solusta verenkiertoon. Infektoituneen solun hajottaminen ja isäntäorganismin makromolekyyliaineenvaihdunnan sulkeminen ovat usein viruksen rakenneproteiinien tehtäviä.
Toista strategiaa käyttävät vaipalliset virukset, joista esimerkkeinä ovat kaikki (-)-juosteen RNA-virukset, togavirukset ja retrovirukset, ja se yhdistää virionin kokoamisen viimeisen vaiheen sen ulostuloon infektoidusta solusta. Näiden vaipallisten virusten tapauksessa asianmukaisia signaalisekvenssejä tai muita tunnistusmarkkereita sisältävät virusproteiinit liitetään plasmakalvon tai muiden sytoplasmisten kalvojen sekä sisä- että ulkopintaan.
Ulkopinnasta ulkonevat proteiinit yleensä glykosyloituvat isäntäentsyymien vaikutuksesta ja aggregoituvat paikkauksiksi, jotka syrjäyttävät isäntäkalvon proteiinit. Viruksen nukleokapsidit sitoutuvat erityisiin viruksen spesifioituihin proteiineihin, jotka vuoraavat näiden paikkausten sytoplasmista puolta, tai virusglykoproteiinien (esim. togavirukset) sytoplasmisiin domeeneihin ja kietoutuvat paikkausten mukana. Prosessissa syntyvä virioni ”pursotetaan” (ekstruusio) solunulkoiseen ympäristöön.
Joissakin tapauksissa (esim. ortomyksovirukset ja paramyksovirukset) yhden pintaproteiinilajin katkaisua ja uudelleenjärjestymistä tapahtuu ekstruusion aikana tai sen jälkeen ja se antaa äskettäin muodostuneelle virionille kyvyn infektoida soluja.
Viruksen kokoaminen ja kypsyminen ekstruusiolla solun pinnalta tarjoaa tehokkaamman ulospääsymekanismin, koska se ei riipu infektoituneen solun hajoamisesta. Itse asiassa virukset, jotka kypsyvät ja poistuvat tällä tavalla, vaihtelevat huomattavasti niiden vaikutuksissa isäntäsolun aineenvaihduntaan ja eheyteen. Ne vaihtelevat erittäin sytolyyttisistä (esim. togavirukset, paramyksovirukset, rabdovirukset) viruksiin, jotka ovat usein ei-sytolyyttisiä (esim. retrovirukset).
Viruksen glykoproteiinien solun pintaan insertoitumisen ansiosta nämä virukset kuitenkin antavat solulle uuden antigeenispesifisyyden ja infektoituneesta solusta voi tulla ja tulee isännän immuunimekanismien kohde. Herpesviruksen nukleokapsidi on koottu tumaan. Toisin kuin muut vaipalliset virukset, vaippa ja kypsyminen tapahtuvat tumakalvon sisälamellilla. Vaipallinen virus kerääntyy tumakalvon sisä- ja ulkolamellien väliseen tilaan, sytoplasmisen retikulumin säiliöihin ja rakkuloihin, jotka kuljettavat viruksen solun pinnalle.
Vaipallinen virus on ainutlaatuisesti suojattu kosketukselta sytoplasman kanssa. Herpesvirukset ovat sytolyyttisiä ja tuhoavat poikkeuksetta solut, joissa ne lisääntyvät. Kuten muut vaipalliset virukset, herpesvirukset antavat tartunnan saaneelle solulle uusia antigeenispesifisyyksiä.
Virusgenomien vaihtelu ja virusten lisääntyminen
Virologian tutkimuksen pääpaino on geneettisten variaatioiden roolissa eri viruslajien sisällä, viallisiin viruksiin sekä rajoittaviin ja abortoiviin infektioihin ihmisten sairauksissa.
Kiinnostus näitä ilmiöitä kohtaan johtuu useista näkökohdista. Näiden joukossa ovat havainnot, joiden mukaan (i) monien ihmisiä tartuttavien virusten aiheuttamien kliinisten sairauksien kirjo vaihtelee huomattavasti vakavuudeltaan ja oireiltaan, (ii) jotkin virukset (esim. ihmisen lentivirukset, influenssa ja muut muut RNA-virukset) mutatoituvat herkästi, ja (ii) että monta vuotta primaaristen infektioiden jälkeen yksilöillä voi esiintyä oireita toistuvista infektioista, keskushermoston kroonisista heikentävistä sairauksista ja pahanlaatuisista kasvaimista, jotka liittyvät tähän primaariseen infektioon.
Ymmärryksemme näiden ilmiöiden keskinäisistä suhteista voidaan tiivistää seuraavaan keskusteluun.
Samaan lajiin ja perheeseen kuuluvat virukset voivat vaihdella suuresti. Esimerkiksi, vaikka epidemiologisesti samankaltaiset ihmisen herpesviruskannat ovat yleensä identtisiä, toisiinsa liittyvät kannat erottuvat helposti restriktioentsyymipolymorfismin avulla. Tämä vaihtelu, niin merkittävä kuin se näyttääkin, hämärtyy havainnolla, että ihmisen immuunikatovirusten peräkkäiset isolaatit voivat erota nukleotidisekvenssistään.
Ajatus siitä, että jotkin luonnollisesti esiintyvät kannat aiheuttavat todennäköisemmin vakavia sairauksia kuin toiset, on enemmän anekdoottinen kuin todistettu, mutta se ei ole kaukaa haettu.
Siirtyessään viruksilla on taipumus tuottaa viallisia mutantteja (variantteja). On kätevää luokitella vialliset virukset kahteen ryhmään.
Ensimmäisen ryhmän viruksilta puuttuu yksi tai useampi välttämätön geeni, ja siksi ne eivät pysty replikoitumaan itsenäisesti ilman auttajavirusta. Kiinnostus tätä ryhmää kohtaan johtuu epäilyistä, että tietyntyyppiset vialliset virukset (esim. papilloomavirukset) voivat muuttaa infektoituneita soluja normaaleista pahanlaatuisiksi tai transaktivoida (esim. herpesvirukset) onkogeenisiä viruksia aiheuttaen solun muuttumisen pahanlaatuiseksi.
Toiseen ryhmään kuuluvat virukset, jotka sisältävät mutaatioita ja deleetioita ja jotka siksi eivät voi replikoitua tehokkaasti. Kiinnostus jälkimmäistä kohtaan johtuu suurelta osin havainnoista, että keskushermoston toimintaa krooniset heikentävät infektiot voivat jollain tavalla liittyä viruksiin. Joidenkin virusten replikaatio on liian hidas ja niiden kyky tuhota tartunnan saaneita soluja tai kyky muuttaa tartunnan saaneita soluja riittävästi tehdäkseen infektoituneesta solusta isäntäorganismin immuunijärjestelmälle kohteen estää sairastuneiden solujen tuhoamisen.
Geenimanipuloidut virukset, joista puuttuu yksi tai useampi geeni ja jotka voidaan luokitella viallisiksi, voivat viime kädessä olla virusten suurin lahja ihmiskunnalle: keino viedä geenejä täydentämään geneettisiä vikoja tai tuhota valikoivasti syöpäsoluja.
Rajoittavat ja abortiiviset infektiot kiinnostavat pääasiassa siksi, että solu voi selviytyä ja säilyttää virusgenomin määräämättömän ajan isännän eliniän ajan. Kompetentilla viruksella (esim. herpesviruksilla) restriktiivisesti infektoitunut solu voi olla viruksen piilevä säiliö, joka voi replikoitua ja levitä, kun solu aktivoidaan sallivaksi. Viallisella viruksella epäonnistuneesti infektoitunut solu voi myös selviytyä ja sopivalla ärsykkeellä siitä voi tulla pahanlaatuinen (esim. papilloomavirukset).
Joissakin tapauksissa rajoittavat infektiot voivat liittyä vaatimukseen, että virusta on säilytettävä tietyssä solussa, jotta se säilyy luonnollisen isäntänsä kanssa. Epäilemättä nämä ilmiöt ovat tutkimuksen kohteena vielä vuosia.
Lähdeaineisto
- Crawford, Dorothy (2011). Viruses: A Very Short Introduction. New York: Oxford University Press. p. 4. ISBN 978-0199574858.
- Cann, Alan (2011). Principles of Molecular Virology (5 ed.). London: Academic Press. ISBN 978-0123849397.
- Scholthof, Karen-Beth G.; Shaw, John G.; Zaitlin, Milton (eds.): Tobacco Mosaic Virus: One Hundred Years of Contributions to Virology. (St. Paul, MN: American Phytopathological Society Press, 1999)
- Calisher, Charles H.; Horzinek, M. C. (eds.): 100 Years of Virology: The Birth and Growth of a Discipline. (New York: Springer, 1999)
- Bos, L. (2000), ’100 years of virology: from vitalism via molecular biology to genetic engineering,’. Trends in Microbiology 8(2): 82–87
- Mateu MG (2013). ”Introduction: The Structural Basis of Virus Function”. Structure and Physics of Viruses. Subcellular Biochemistry. 68. Nature Public Health Emergency Collection. pp. 3–51. doi:10.1007/978-94-007-6552-8_1. ISBN 978-94-007-6551-1. PMC 7120296. PMID 23737047.
- Themes, U. F. O. (2017-02-19). ”6 Viruses–Basic Concepts”. Basicmedical Key. Retrieved 2020-05-29.
- ”Prion Diseases”. CDC. Archived from the original on 2010-03-04. Retrieved 2016-03-25.
- Evans, Alfred (1982). Viral Infections of Humans. New York: Plenum Publishing Corporation. pp. xxv–xxxi. ISBN 0306406764.
- Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F (July 2014). ”Reactivated herpes simplex infection increases the risk of Alzheimer’s disease”. Alzheimer’s & Dementia. 11 (6): 593–99. doi:10.1016/j.jalz.2014.04.522. PMID 25043910. S2CID 28979698.
- ”1 Moved | Prion Diseases | CDC”. Archived from the original on 2010-03-04. Retrieved 2017-09-17.
- Dimmock NJ, Easton AJ, Leppard K, Introduction to Modern Virology, (Oxford: Blackwell Publishers, 2007), ch 23 ”Horizons in human virology”, subch 23.3 ”Subtle and insidious virus-host interactions”, sec ”Virus infections can give their host an evolutionary advantage”, p. 432.
- Stanley Maloy. ”Horizontal Gene Transfer”. Retrieved 2016-03-25.
- Viruses: The new cancer hunters, IsraCast, 1 March 2006
- Castro, Christina; Marine, Rachel; Ramos, Edward; Ng, Terry Fei Fan (22 June 2020). ”The effect of variant interference on de novo assembly for viral deep sequencing”. BMC Genomics. 21 (1): 421. doi:10.1186/s12864-020-06801-w. PMC 7306937. PMID 32571214.
- ”Virus”, Merriam-Webster, Inc, 2011.
- Sussman, Max; Topley, W. W. C.; Wilson, Graham K.; Collier, L. H.; Balows, Albert (1998). Topley & Wilson’s microbiology and microbial infections. London: Arnold. p. 3. ISBN 0-340-66316-2.
- Iwanowski, D. (1892). ”Über die Mosaikkrankheit der Tabakspflanze”. Bulletin Scientifique Publié Par l’Académie Impériale des Sciences de Saint-Pétersbourg / Nouvelle Serie III (in German and Russian). St. Petersburg. 35: 67–70. Translated into English in Johnson, J., Ed. (1942) Phytopathological classics (St. Paul, Minnesota: American Phytopathological Society) No. 7, pp. 27–30.
- Iwanowski, D. (1903). ”Über die Mosaikkrankheit der Tabakspflanze”. Zeitschrift für Pflanzenkrankheiten und Pflanzenschutz (in German). 13: 1–41.
- Pennazio S (2007). ”Genetics and virology: Two interdisciplinary branches of biology”. Rivista di Biologia. 100 (1): 119–46. PMID 17592822.
- Van Epps HL (2005). ”Peyton Rous: Father of the tumor virus” (PDF). Journal of Experimental Medicine. 201 (3): 320. doi:10.1084/jem.2013fta. PMC 2213042. PMID 15756727.
- The Medical and Scientific Conceptions of Influenza, Human Virology at Stanford
- Montagnier L (2002). ”Historical essay. A History of HIV Discovery”. Science. 298 (5599): 1727–28. doi:10.1126/science.1079027. PMID 12459575. S2CID 57481800.
- Gallo RC (2002). ”Historical essay. The Early Years of HIV/AIDS”. Science. 298 (5599): 1728–30. doi:10.1126/science.1078050. PMID 12459576. S2CID 82899411.
- Gallo RC, Montagnier L (2002). ”Historical essay. Prospects for the Future”. Science. 298 (5599): 1730–31. doi:10.1126/science.1079864. PMID 12459577. S2CID 34227893.
- Hu, Wenhui; Kaminski, Rafal; Yang, Fan; Zhang, Yonggang; Cosentino, Laura; Li, Fang; Luo, Biao; Alvarez-Carbonell, David; Garcia-Mesa, Yoelvis (2014-08-05). ”RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection”. Proceedings of the National Academy of Sciences. 111 (31): 11461–66. Bibcode:2014PNAS..11111461H. doi:10.1073/pnas.1405186111. ISSN 0027-8424. PMC 4128125. PMID 25049410.
- 2000 Albert Lasker Award for Clinical Medical Research Archived October 28, 2007, at the Wayback Machine, The Lasker Foundation. Accessed 20 February 2008
- Debyser Zeger (2003). ”A Short Course on Virology / Vectorology / Gene Therapy” (PDF). Current Gene Therapy. 3 (6): 495–99. doi:10.2174/1566523034578122. PMID 14683447. Archived from the original (PDF) on 2008-08-02.
- Kolata, Gina (2005-10-06). ”Experts Unlock Clues to Spread of 1918 Flu Virus”. The New York Times. ISSN 0362-4331. Retrieved 2008-02-03.
- Stem Cells – This Time without the Cancer, Scientific American News, 30 November 2007
- ”Biggest Known Virus Yields First-Ever Virophage”. Microbe Magazine. November 2008. Archived from the original on July 22, 2011.
- ”Virus hiding in our genome protects early human embryos”. New Scientist. 20 April 2015.
- H. Tang et al., Human Organs-on-Chips for Virology, Trends in Microbiology (2020)https://en.wikipedia.org/wiki/Virology
https://www.ncbi.nlm.nih.gov/books/NBK8181/