Aineenvaihdunnan, inflammaation, kognition ja yleisen terveyden modulointi ketogeenisellä ruokavaliolla
Vähähiilihydraattinen ja runsasrasvainen ketogeeninen ruokavalio (LCHF), on menneiden vuosikymmenten saatossa nostettu tuon tuosta esiin terapeuttisena vaihtoehtona metabolisen oireyhtymän, ylipainon ja lihavuuden sekä eräiden lääkeresistenttien sairauksien, kuten epilepsian, syövän, dementian ja masennuksen hoitona. Oma motiivini selvitellä näitä on se, että ketogeenisen ruokavalion neuroprotektiivinen ja tulehduksia hillitsevä luonne voi hidastaa etenevään MS-tautiin liittyvien keskushermoston vaurioiden kehittymistä.
Ruokavaliota on hyödynnetty lääkehoidon rinnalla tai lääkehoidosta riippumatta vuosisatoja. Esimerkiksi diabeteksen hoitoon suositeltiin vähähiilihydraattista ruokavaliota jo 1700-luvun lopulla.
Tutuin tehokkaan ravintoterapian kohde on keliakia, jota sairastavat voivat elää jokseenkin normaalia elämää välttämällä viljojen sisältämää gluteenia. Lääkeresistenttiin epilepsiaan ei edelleenkään tunneta parempaa hoitoa, kuin ketogeeninen ruokavalio, jota on käytetty erityisesti lasten epileptisten kohtausten hillitsemiseen 1920-luvulta alkaen.
Tämän ruokavalion kiistattomista hyödyistä huolimatta, terveydenhuollon ja ravitsemuksen ammattilaiset kyseenalaistavat yhä ketogeenisen ruokavalion turvallisuuden sen aiheuttamien kohonneiden seerumin ketoaineiden ja ruokavalion rajoitetun ravintokuitujen saannin vuoksi.
Ruokavalion herättämiä epäilyjä lisää edelleen huoli aivojen glukoosinsaannin riittävyydestä sekä tyydyttyneisiin rasvoihin ja kolesteroliin liittyvät irrationaaliset pelot.
Siirtymävaiheessa ketogeeninen ruokavalio voi aiheuttaa energiasubstraatin vaihtumisen ja nestehukan seurauksena ohimenevän ketoflunssan. Se on tavallista, eikä lainkaan vaarallista. Usein se kertoo, että ruokavaliomuutoksen jälkeen vettä pitäisi juoda enemmän, koska sokereiden rajoittaminen poistaa kehosta nesteitä.
Ketogeeninen ruokavalio on turvallinen ja tehokas terapiavaihtoehto moniin aineenvaihduntasairauksiin. Tässä katsauksessa tutustutaan eksogeenisten ketoaineiden ja ketonilähteiden aineenvaihduntahyötyjen tieteellisiin perusteisiin.
Katsauksessa käsitellään myös eksogeenisen β-hydroksibutyraatin (BHB) ja siihen liittyvän lyhytketjuisen rasvahapon, butyraatin (BA), synergiaa (yhteisvaikutusta) solutason aineenvaihduntatapahtumissa.
β-hydroksibutyraatin ja butyraatin hyödyt aineenvaihdunnan, inflammaation, kognition ja yleisen terveyden moduloinnissa
Monet soluistamme voivat käyttää rasvahappoja ATP-tuotannon energiasubstaattina, jos glukoosia ei ole riittävästi saatavilla. Aivot eivät kuitenkaan voi suoraan hapettaa rasvohappoja energiaksi, koska rasvahapot eivät läpäise veri-aivoestettä. Vesiliukoinen pienemmän molekyylipainon omaava ketoaine läpäisee vaivatta veri-aivoesteen ja tarjoaa hermosoluille erittäin tehokkaan energialähteen [1, 2].
Ketoaineet, kuten β-hydroksibutyraatti, ovat neuroneille erinomaisia energiasubstraatteja. Erityisen tärkeitä ketoaineet ovat henkilöille, joiden hermosolujen glukoosimetabolia (solujen glukoosin otto) on heikentynyt geneettisten tai elintapoihin liittyvien syiden vuoksi [3]. Ketoaineet aktivoivat mm. kognitiivisista häiriöistä kärsivien aivosolujen energiantuotantoa [4, 5].
Ruokavalion sisältämällä rasvalla on väitetty olevan ratkaiseva rooli ihmisen aivojen evoluutiossa, koska aivot tarvitsevat runsaasti energiaa sisältävää ravintoa sekä rasvojen sisältämiä rakennuspalikoita [6] ja kolesterolia. Tällaista käsitystä tukee huomio, joka osoittaa, että dokosaheksaeenihapolla (DHA) ja muilla rasvoilla on ratkaiseva rooli hermokudosten kasvussa ja toiminnassa. Rasva-aineenvaihdunnan poikkeavuudet tai ravintorasvojen puutteet voivat häiritä aivojen kehitystä ja toimintaa [7].
Eräät asiantuntijat arvelevat, että siirtyminen runsasrasvaisesta ruokavaliosta vähärasvaiseen ruokavalioon on selittävä syy Pohjois-Amerikan metabolisen oireyhtymän (insuliiniresistenssi, diabetes, verenpaine, dyslipidemia, lihavuus) yleistymisen taustalla. USAn makroravinteiden kulutuksen tilastollinen tarkastelu osoittaa lihavuuden lisääntymisen korreloivan ravinnon rasvan vähentämisen kanssa. Rasvan kulutuksen vähentäminen on puolestaan lisännyt runsaasti hiilihydraatteja (sokereita) sisältävien ruokien kulutusta [8].
Samalla noususuuntaisella tilastokäyrällä ovat vuoden 1980 jälkeen kolminkertaistunut lihavien määrä ja aikuistyypin diabeetikkojen määrän kaksinkertaistuminen samana aikana. Iltapäivälehtien clickbait-jutut ketogeenisellä ruokavaliolla sairastuneista kannattaa jättää omaan arvoonsa. Diabeetikkojen määrä on globaalisti jo lähes puoli miljardia ja lihavia on kolmannes kaikista ihmisistä.
Iltapäivälehtien tulisi kiinnittää huomiota todelliseen ongelmaan: Suomessa on puoli miljoonaa aikuistyypin diabetesta sairastavaa. Näistä tilastollisesti joka toinen tulee kuolemaan sydän- ja verisuonitauteihin.
Kaiken lisäksi diabeteksen hoitokustannukset Suomessa ovat samaa luokkaa tai korkeammat kuin tupakoinnin ja alkoholin aiheuttamien sairauksien hoitokustannukset. Koskettavat mielipiteitä muokkaavat tarinat ketogeenisellä ruokavaliolla elämänsä tuhonneesta Penasta tai Sirkka-Liisasta eivät muuta tosiasioita miksikään: voi ja kolesteroli eivät ole suomalaisten suurin terveysongelma.
Tämän hetken kriittisin terveysongelma on hyperglykemian ja hyperinsulinemian aiheuttama insuliiniresistenssi sekä siihen liittyvät aineevaihduntasairaudet. Niiden hoito ravintoterapialla on helppoa ja halpaa.
Jossain ruokavalioiden ääripäiden välillä voi olla terveyden Shangri-La, jossa jalostettuja hiilihydraatteja (sokereiden lähteitä) rajoitetaan, tyydyttyneitä rasvoja ei pelätä ja tuoreilla (matalan glykeemisen indeksin) vihanneksilla on edelleen tärkeä rooli osana terveellistä ruokavaliota [9]. Tai sitten sellaista ei ole.
Energiansaannin rajoittaminen paastoamalla tai ruokavalion sisältämien hiilihydraatteja rajoittamalla johtaa ketoosiin ja seerumin ketonipitoisuuden nousuun [10].
Ketogeeninen vähähiilihydraattinen, runsasrasvainen ruokavalio (LCHF) on kokenut kuluneiden sadan vuoden aikana monta renesanssia ja romahdusta. Jotkut, jotka eivät tunne historiaa, pitävät ketogeenistä ruokavaliota vain muotioikkuna (fad), mutta hiilihydraattien rajoittamista on harjoitettu terveyden kohentamiseksi jo esikristillisillä ajoilla. Lähes jokaiseen uskontoon sisältyy puhdistava paasto, eikä se ole sattumaa, sillä paastolla on tunnustettuja terveyshyötyjä. Paasto johtaa ketoosiin.
Viime vuosisadalla ketogeenisen ruokavalion positiivisista terveysvaikutuksista raportoitiin laajemmin esimerkiksi 1930- ja 1940-luvuilla, jolloin sitä hyödynnettiin mm. astman hoidossa.
Ketogeenistä ruokavaliota on käytetty tehokkaasti hoitona:
-
metaboliseen oireyhtymään[11]
-
epilepsiaan [12]
- kognitiivisten ja neurologisten häiriöiden [13], kuten Alzheimerin taudin hoitona, jossa sen on osoitettu vähentävän haitallista amyloidiproteiinia [14]
-
termogeneesin proteiiniaktiivisuuden irrottamisen aktivaattorina [15]
-
laihduttamiseen [16]
Ketogeeninen ruokavalio ei ole uusi ja muodikas ruokavalio-oikku, vaan ruokavalio, johon kehomme on täydellisesti adaptoitunut nisäkkäiden ja hominidien evoluution aikana.
Se, että tämä ruokavalioprotokolla voi tehokkaasti vähentää epileptisten kohtausten esiintymistiheyttä [17] ja auttaa hoitamaan lääkeresistenttiä epilepsiaa [18], vahvistettiin jo 1920-luvulla [19, 20].
Tässä katsauksessa käydään läpi joitain ketogeenisen ruokavalion metabolisten ja terveydellisten hyötyjen todisteita, sekä tarkastellaan ruokavalion turvallisuutta ja tehoa terapiavaihtoehtona lääkkeiden rinnalla ja lääkkeistä riippumatta.
Tieteellinen näyttö esitetään myös eksogeenisten ketoaineiden ja muiden erityyppisten ketonilähteiden antamiselle hiilihydraatteja rajoittavan ruokavalioprotokollan täydennyksenä tai vaihtoehtona ruokavaliolle.
Kirjoittajat suosittelevat erityistä menettelytapaa, johon sisältyy eksogeenisen ketonin, β-hydroksibutyraatin (BHB) antaminen lyhytketjuisen rasvahapon, butyraatin (BA) mukana.
Tässä katsauksessa painotetaan tämän BHB-BA-yhdistelmän synergiaa solusignaloinnin ja elimistön hiljaisen tulehduksen, eli inflammaation hallinnan yhteydessä ja sen käyttöä energiasubstraattina ATP: n muodostamiseen TCA-syklissä (sitruunahappokierrossa).

2. Mitä ketogeenisellä ruokavaliolla tarkoitetaan?
Ketogeenisessä ruokavaliossa ravintoaineiden makroravinnprofiili on tärkeä. Päivittäinen energiansaanti sisältää:
-
65–70% rasvaa
-
20% proteiinia
-
5–10% hiilihydraatteja
Ketogeeninen ruokavalio kääntää perinteisen ravintopyramidin ylösalaisin. Päivittäinen hiilihydraattien saanti, joka ei ylitä 75 grammaa, on vähimmäisedellytys ketoosissa pysymiseen; 50 gramman hiilihydraattien saannin enimmäismäärä on toki ketoosin ylläpitämisen kannalta turvallisempi hiilihydraattien saanti. Ketogeenisen ruokavalion alussa hiilihydraattien saantia voi olla järkevää rajoittaa ~20 grammaan päivässä, ja monet ketoilijat pysyvät ~20 gramman päiväsaannissa ilman mitään ongelmia.
Mitä vähämmän hiilihydraatteja ravinto sisältää, sitä tehokkaammin elimistö purkaa rasvasolujen sisältämiä triglyseridejä verenkiertoon, tuottaa ketoaineita energiasubstraateiksi ja hapettaa vapaita rasvahappoja betaoksidaatiossa.
Ketogenressä 75 grammaa hiilihydraatteja päivässä on jo melkoisen villiä sokerihurjastelua, mutta virallinen linja, josta olen kuullut puhuhttavan, on, että alle 150 grammaa hiilihydraatteja päivässä luokitellaan vähähiilihydraattiseksi ruokavalioksi tai karppaamiseksi. Sellainen on absurdia roskaa.
Minä en laske sen enempää hiilihydraatteja, kuin kaloreita. Syön ravintoa, jossa on hiilihydraatteja vähän (alle 6 g/ 100 grammassa) tai ei ollenkaan. Hiilihydraattien saanti vaihtelee minulla keskimäärin 20 ja 50 gramman välillä päivässä. 50 gramman ylittäminen näkyy painossa, verenpaineessa ja verensokerissa. Se ei sovi minulle. Joillekin 50-100 g hiilihydraatteja päivässä voi sopia.
Annos keitettyä riisiä sisältää ~50 gramman hiilihydraatteja. Suuri omena tai banaani, joissa hiilihydraattien määrä on ~40 grammaa, voivat katkaista ketoosin, etenkin kun päälle lasketaan muut päivittäiset hiilihydraattien lähteet.
Myös ruokavalion sisältämillä proteiineilla on vaikutusta seerumin glukoosipitoisuuteen. Esimerkiksi leusiinilla jota saadaan yleensä riittävästi arkiruoasta (eläinperäisestä ravinnosta sekä palkokasveista, siemenistä ja hiivasta), voi olla merkittävä vaikutus ketogeneesin aktivointiin, insuliiniherkkyyteen ja veren puhdistamiseen glukoosista [21].
Sen sijaan eräät mut aminohapot, kuten alaniini, kysteiini ja glysiini, ovat erittäin glukoneogeenisiä (ts. glukoneogeneesiä indusoivia). Matalan energiansaannin aikana keho voi helposti syntetisoida glukoneogeenisiä aminohappoja glukoosiksi [22]. Glukoneogeenisiin / glukogeenisiin aminohappoihin kuuluvat myös arginiini, seriini ja proliini.
Jos ravinto sisältää runsasti glukoneogeenisiä aminohappoja, niistä tuotetaan glukoneogeneesissä glukoosia, mikä kohottaa verensokeria ja insuliinipitoisuutta ehkäisten ketogeneesin käynnistymistä.
Vaikka kohtalaiseen hyperketonemiaan liittyy merkittäviä terveysvaikutuksia riippumatta siitä, käytetäänkö sitä ravintoterapiana tai yksinkertaisesti elämänlaadun parantamiseen, tätä tilaa ei ole helppo saavuttaa ja ylläpitää ilman suunnittelua ja ruokavaliossa tehtäviä uhrauksia [23, 24]. Itse asiassa ketogeenistä elämäntapaa on nykyään jo hieman hankala ylläpitää, kun otetaan huomioon hiilihydraatti- ja sokerikeskeinen kulttuurimme. Hiilihydraattien lähteet ovat hyvin piilossa monissa arkisissa ja jalostetuissa elintarvikkeissa. Moni ei esimerkiksi tule ajatelleeksi, että maito sisältää sokeria (maitosokeria, eli laktoosia).
Yhtäältä lääketieteellisen yhteisön ketogeeniseen ruokavalioon ja varsinkin ketoasidoosiin liittyvä virheellinen viestintä ohjaa väestön kulutustottumuksia kohti hiihihydraattien runsasta saantia.
Ketoasidoosi ja ketoosi sotketaan iloisesti keskenään. Ravintoketoosi on kuitenkin hyvin erilainen fysiologinen tila kuin ketoasidoosi.
Hiilihydraattien rajoittamiseen tai paastoon perustuvista ruokavalion muutoksista johtuva ketoosi ei tarkoita samaa kuin tyypin 1 diabetekseen ja siihen liittyviin diabeettisiin tiloihin liittyvä patologinen ketoasidoosi [25, 26]. Turvallinen hyperketonemia voi saavuttaa jopa 10 mmol/l ketoaine-pitoisuuden paastoamalla tai ketogeenisella ruokavaliolla [27, 28]. Keho säätelee ketoosia autonomisilla palautemekanismeilla [29]. Ketoasidoosille on ominaista seerumin ketonitasot, jotka ylittävät 18 mmol/l [30].
| Ketoasidoosi on fysiologinen tila, jossa jotkin solujen ulkoiset nesteet happamoituvat kun niihin kertyy liikaa happamia ketoaineita. Ihmisillä ketoasidoosit jaetaan aiheuttajien mukaan muun muassa diabeettiseen ketoasidoosiin (DKA) ja alkoholiketoasidoosiin (AKA). Yleisempi diabeettinen ketoasidoosi voi johtaa hoitamattomana kuolemaan. Happomyrkytys on hengenvaarallinen tila, mutta aivan eri eri asia kuin terveen ihmisen paastotessa muodostuvat ketoaineet eli nälkähapot(diabetes.fi). Diabeetikon uhkaavasta happomyrkytyksestä kertoo se, kun verensokeri on koholla ja samaan aikaan verestä löytyy ketoaineita. DKA:n ja AKA:n yhteisiä oireita ovat muun muassa hyperventilaatio, oksentelu, mahakipu, sydämen tiheälyöntisyys ja matala verenpaine. Usein DKA:ssa ilmenee korkea verensokeri, potilas on sekava ja hengitys haisee asetonilta (hedelmäiseltä). Verensokeritaso on AKA:ssa usein normaali tai matala, potilas on lähes tajuissaan ja hengitys ei juurikaan haise asetonilta. – Wikipedia & Diabetes.fi |
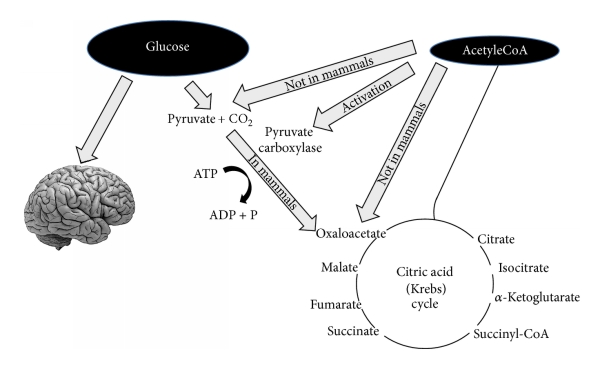
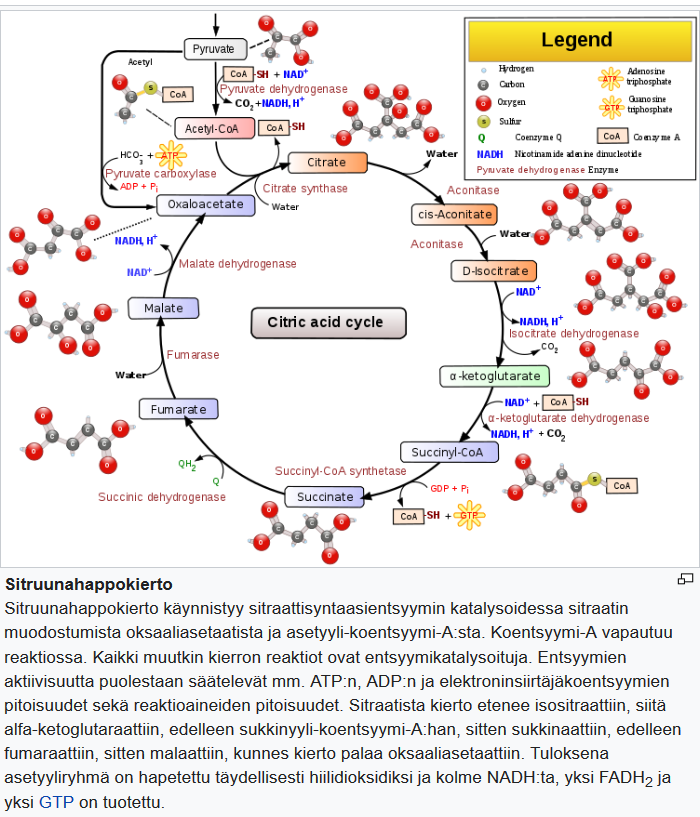
Koska ketogeeninen ruokavalio muuttaa kehon energia-aineenvaihduntaa glukoosipolttoisesta rasvapolttoiseksi, se imitoi paastoa. Ketogeenisen ruokavalion vaikutukset aineenvaihdunnan modulointiin ovat samanlaisia kuin paaston vaikutukset. Solujen energiasubstraatti vaihtuu glukoosista ketoaineiksi ja vapaiksi rasvahapoiksi, joista hapetetaan asetyylikoentsyymi-A:ta sitruunahappokiertoon.
Energiasubstraatin muutos käynnistää solujen puhdistusjärjestelmän, eli autofagian, joka siivoaa soluja kuona-aineista ja tuottaa niistä energiaa.
Miksi ketogeeninen elämäntapa?
Nykyisillä elintavoilla monet syövät huomamattaan aivan liian hiilihydraattipainotteisesti. Hiilihydraatit muodostuvat sokereista ja kohottavat seerumin glukoosi- ja insuliinipitoisuuksia.
Glukoosi ja sen pitoisuuden kasvun lisäämä seerumin insuliini vaurioittavat esidiabeettisella ja diabeettisella tasolla etenkin kehon pieniä verisuonia. Diabeteksen kehittymisen voi välttää tarkkailemalla sokereiden saantia.
Maksassa ylimääräinen glukoosi (ja fruktoosi) muutetaan lipogeneesissä triglyserideiksi (vrt. alkoholista riippumaton rasvamaksa). Verenkiertoon maksasta erittynyt ylimääräinen glukoosi varastoidaan ylimääräisen rasvan tapaan rasvasoluihin, jossa se muutetaan de novo lipogeneesissä triglyserideiksi.
Ylimääräinen glukoosi on siivottava verenkierrosta, koska glukoosi glykatoituu veressä olevien muiden molekyylien kanssa. Glykaation lopputuotteet (AGE) altistavat monille sairauksille. Tämä on myös se syy, miksi diabetes lisää virtsaamistarvetta: keho yrittää pissaamalla päästä eroon ylimääräisistä sokereista.
Ketogeeninen ruokavalio ei aiheuta ketoasidoosia terveillä. Jatkuvasti kohollaan oleva verensokeri ja korkea insuliini kasvattavat metabolisen oireyhtymän ja insuliiniresistenssin (ne ovat itse asiassa sama asia) ja diabeteksen riskiä. Tyypin 2 diabetes aiheuttaa lihavuutta, alkoholista riippumatonta rasvamaksaa sekä sydän- ja verisuonitauteja monien muiden aineenvaihduntaan kytkeytyvien sairauksien lisäksi.
Tyypin 2 diabetes on ongelma ja ketogeeninen ruokavalio ongelman ratkaisu.
Kun seerumin glukoosia hoidetaan väärin, seurauksena olevat edistyneet glykaation lopputuotteet (AGE) [31, 32] sekä inflammaatio [33, 34] aiheuttavat merkittävää veren toksisuutta [35] ja lisäävät sairastumisriskiä [36].
Glykaation kehittyneille lopputuotteille (AGE) altistunut LDL (matalatiheyksinen lipoproteiini) on ateroskleroosin ja muiden sydän- ja verisuonitautien riskitekijä ja aiheuttaja [37].
LDL itsessään ei ole sydän- ja verisuonitautien riskitekijä, vaan elimistön luonnollinen triglyseridejä, kolesterolia ja rasvaliukoisia vitamiineja kuljettava kuljetusmolekyyli (lipoproteiini), mutta korkean verensokerin aiheuttaman glykaation ja vapaiden happiradikaalien hapettamasta LDL-partikkelista tulee sydäntautien riski.
Elimistön reaktio (hyperglykemia, hyperinsulinemia, glykaatio ja insuliiniresistenssi) seerumin korkeaan glukoosiin, jonka aiheuttaa korkean glykeemisen kuorman ravinto, ei ole terveyttä ja kehon hyvinvointia edistävä. Monet meistä myrkyttävät itseään sokereilla.
Endokriinisen fysiologian peruskäsitys kertoo meille, että joka kerta kun insuliinia erittyy haimasta korkean glykeemisen kuorman ruokien saannin vuoksi tai sitä piikitetään haiman vaurioituneen insuliinintuotannon vuoksi kohonneen glukoosin laskemiseksi, lipolyysi estyy ja energiasubstraatit, glukoosi ja rasvahapot varastoidaan [38]. Tämä toiminta lisää rasvan kertymistä erityisesti sisäelinrasvana ja viskeraalisena keskivartalolihavuutena, mikä vähentää rasvahappojen syntetisoimista ketoaineiksi tai hapettumista betaoksidaatiossa.
Seerumin ketoaineiden saanti soluissa tapahtuu insuliinista riippumattomien metabolisten reittien kautta [39]. Siksi, vaikka insuliiniresistenssi heikentää glukoosin ottoa insuliinista riippuvaisissa soluissa, ketoaineita voidaan hyödyntää energiasubstraatteina insuliinin aineenvaihduntahäiriöistä huolimatta.
Tämä on valtava kehitysaskel neurodegeneratiivisten sairauksien, kuten Parkinsonin ja Alzheimerin taudin tulevia terapiavaihtoehtoja suunniteltaessa. Glukoosin heikentynyt energiametabolia aivoissa on yksi, ei toki ainoa, tekijä monitekijäisissä neurodegeneratiivisissa sairauksissa.
Lisäksi todisteet osoittavat, että kohonneet seerumin ketoainepitoisuudet vähentävät maksan glukoosintuotantoa ja auttavat tällä mekanismilla myös lieventämään kohonneita seerumin glukoosipitoisuuksia [40].
Ketogeeninen ruokavalio on tehokkain lääkkeetön hoito tyypin 2 diabetekseen, metaboliseen oireyhtymään ja alkoholista riippumattomaan rasvamaksaan. LCHF voi kääntää alkavan diabeteksen suunnan [41] ja johtaa aikuistyypin diabeteksen lääkkeettömään remissioon. Hiilihydraattirajoitus vaikuttaa tehokkaasti painonhallintaan [42, 43], laskee seerumin glukoosia eli verensokeria prediabeettisilla sekä diabetesta sairastavilla potilailla [44]. Ketogeeninen ruokavalio laskee myös insuliinin tarvetta insuliiniriippuvaisissa aikuistyypin diabeettisissa oireissa [45, 46].
Hiilihydraattirajoitus ei ole ainoa ruokavaliostrategia, joka torjuu elämäntapaan liittyviä sairauksia. On monta tapaa syödä oikein ja vähintään yhtä monta tapaa syödä väärin.
Ketogeeninen ruokavalio on kuitenkin yksi tehokkaimmista solujen ja elimistön hyvinvointia ylläpitävistä ruokavalioista. Niin hyödyllinen kuin se onkin painonhallinnassa ja metabolisen oireyhtymän terapiana, ketogeenisellä ruokavaliolla tapahtuva kalorirajoitus on tunnetusti huonosti siedetty, ellei sitä kompensoida korkeammalla rasvasta saadulla energialla [47]. Riittävästi rasvaa sisältävä ruoka pitää nälän tehokkaasti loitolla ja ravinnon energiapitoisuus laskee kaloreita miettimättä.
LCHF-ruokavalio myötävaikuttaa seerumin glukoosin ja paastoseerumin glukoosin laskuun sekä parantaa glukoositoleranssia [48]. Jos hiilihydraattien saanti on riittävän matala, seerumin ketonitasot voivat kasvaa riittävästi täyttämään elimistön energiantarvetta ja tukemaan terveyttä useilla tavoilla [49, 50].
Silti vähähiilihydraattisen ruokavalion edellyttämien uhrauksien, kuten leivästä, perunasta, pizzasta, hampurilaisista, bissestä ja sokeriherkuista luopumisen vaikeus on ketoilijoille haaste, joka johtaa herkästi ketogeenisestä ruokavaliosta luopumiseen.
Tämä on hyvin tavallista ruokavalion alkuvaiheessa, mutta vähitellen kaikki sokeriin liittyvät mielihalut vain katoavat. Rasva pitää nälän erinomaisesti loitolla ja energiatasot pysyvät vahvoina koko päivän 1-3 aterialla ilman parin tunnin välein mussutettavia välipaloja.
Monille meistä lääkärin määräämä pilleri tai dosetillinen päivän käynnistäviä lääkkeitä voi olla kuitenkin helpompi ratkaisu, kuin hieman selkärankaa ja sokereista luopumista edellyttävä ketogeeninen ruokavalio.

3. Endogeenisten ketoaineiden muodostuminen
Lihomisen ja laihtumisen metabolinen perusta
Lipolyysi purkaa rasvasoluihin varastoituja triglyseridejä vapaiksi rasvahapoiksi ja glyseroliksi verenkiertoon lipolyyttisten hormonien (glukagoni, kortikotropiini, adrenaliini ja noradrenaliini) vaikutuksesta.
Veren insuliinipitoisuus säätelee lipolyyttisten hormonien erittymistä. Insuliini on myös lipolyysin tarvitsemien entsyymien estäjä, joten, kun veren insuliinipitoisuus on korkea, lipolyysi ei voi käynnistyä.
Käytännössä: Kaloreita rajoittavalla dieetillä, jossa suuri osa päivittäisestä energiasta otetaan hiilihydraateista, rasvasolujen polttaminen energiaksi estyy veren jatkuvasti korkean insuliinipitoisuuden vuoksi. Tämä tarkoittaa sitä, että painon laskua tapahtuu lähinnä rasvattoman massan (lihasten) vähenemisen kautta. Niukkakalorisella hiilihydraattipitoisella dieetillä rasvaa poltetaan yöaikaan, sillä insuliinipitoisuus laskee riittävästi ~8 tuntia syömisen jälkeen, ja vasta silloin lipolyysi voi käynnistyä. Tällöin laihtumisen aikaikkuna jää kuitenkin verrattain lyhyeksi.
Lipolyysin vastareaktio on lipogeneesi, joka edistää insuliinin vaikutuksesta rasvan ja sokereiden varastoimista rasvasoluihin triglyserideinä. Evoluution ja aineenvaihdunnan kannalta lihominen on perusteltua vain, jos rasvasoluihin tallennettu energia voidaan hyödyntää energiaksi silloin, kun ravinnosta saadaan puutteellisesti energiaa. Tämä on lihomisen ja laihtumisen metabolinen perusta.
Maksa on rasvasoluista vapautuneiden rasvahappojen ja glyserolin ensisijainen kohde. Ketoaineita tuotetaan vapaista rasvahapoista maksassa tapahtuvassa ketogeneesissä. Ketoaineet voivat myöhemmin toimia aivojen energiasubstraateina [51–53].
Triglyseridien glyseroliosaa käytetään glukoosia syntetisoivassa glukoneogeneesissä. Keho pystyy helposti syntetisoimaan kaiken tarvitsemansa glukoosin. Sanonpahan vain, koska kymmenen vuotta sitten aiheesta väiteltiin ankarasti.
Terveellä ihmisellä, jolla haiman β-solut toimivat normaalisti, seerumin ketonitasoja hallitaan autonomisesti [54]. Seerumin ketonit, asetoasetaatti ja asetoni, samoin kuin β-hydroksibutyraatti, toimivat signaaliligandeina, jotka säätelevät maksan β-oksidaatiota [55] seerumin ketoaineiden kuormituksen säätelemiseksi.
Vuosikymmenien aikana on kerääntynyt kiistattomia todisteita, jotka tukevat tämän palautejärjestelmän olemassaoloa ja tehokkuutta sekä ketonisynteesin huolellista säätelyä transkriptiotasolla [27].
Kuinka seerumin ketonikertomus liittyy seerumin glukoosimalliin?
Tarina on rinnasteinen. Molemmissa malleissa huonosti säännellyt energiasubstraatin tasot voivat johtaa toksiseen tasoon, mutta tasot, joita terve fysiologia hallitsee autonomisesti, tukevat terveellistä aineenvaihduntaa.
Itse asiassa terve seerumin glukoosipitoisuus (~5,0 mmol/l) ei ole sen ihmeellisempi kuin seerumin ketonien terveellinen taso. Seerumin ketonien terveeksi tasoksi hyperketonemian yhteydessä on dokumentoitu 2,0 mmol/l – 8,0 mmol/l [56].
Tätä ketoositasoa pidetään lievänä tai kohtalaisena hyperketonemiana, jonka keho tuottaa selviytymismekanismina pitkittyneisiin paastojaksoihin [25, 53].
Ketoaineiden perustaso terveillä henkilöillä vaihtelee 0,1 – 0,2 mmol/l pitoisuutena [57]. Seerumin ketoneja käytetään useimmissa kudoksissa tehokkaasti energiasubstraateina silloin kun glukoosia on niukasti saatavilla [58]. Ketoaineita käyttävät mm.sydän [59] ja aivot. Sydän on hyvin joustava energiasubstraattien suhteen, mutta sydämen energiansaannin kannalta tehokkain energiasubstraatti on rasva, joka hapettuu β -oksidaatiossa. Sydänlihaksen soluihin varastoituu herkästi lipotoksiineja, jos veren glukoosi- ja rasvapitoisuus on jatkuvasti liian korkea ja sydänlihakselle syötetään liikaa erilaisia energiasubstraatteja. Sydänkin rasvoittuu.
Tästä rajoittavasta ruokavaliosta voidaan tehdä siedettävämpi antamalla eksogeenistä ketonilisää (lisäravinteena) etenkin, jos halutaan kiihdyttää ketoosin positiivisia metabolisia vaikutuksia elimistössä. Endogeenisen (elimistön tuottaman) ketoosin käynnistymisen aikaikkuna ~20 g päivittäisillä hiilihydraateilla on 2-3 vuorokautta [60, 61].
Eksogeenisten ketoneiden ja erityisesti β-hydroksibutyraatin tutkimus lääkinnällisenä ja elimistön toimintaa tehostavana metabolisena substraattina on hyvin aktiivista. β-hydroksibutyraatin vaikutuksia tutkitaan aiemmin mainittujen neurodegeneratiivisten sairauksien terapian lisäksi NASAn rahoittamana astronauttien kognitiivisten kykyjen parantamiseksi äärioloissa ja USAn puolustusministeriön rahoittamana taistelusukeltajien toimintakyvyn tehostamiseksi ja sukellusaikojen pidentämiseksi.
Ketogeenisen ruokavalion aloittamisen jälkeen seerumin ATP-tuotannon kannalta riittävän ketonitason saavuttaminen, voi kestää hiilihydraattien saannista riippuen jopa viisi päivää (yleensä 2-3 päivää). Nämä siirtymäpäivät voivat osoittautua vaikeiksi ja johtaa huijauspäiviin. Seerumin toiminnallisten ketonitasojen ylläpito edellyttää ruokavalion noudattamisesta [62, 63]. Tässä eksogeeninen ketonilisäaine voi helpottaa ketogeeniselle ruokavaliolle siirtymistä.
Huijaaminen ketogeenisen ruokavalion aikana hidastaa aineenvaihdunnan siirtymistä glukoosimetaboliasta rasvametaboliaan, ketogeneesiin ja β-oksidaatioon, joka itse asiassa on ketogeenisen ruokavalion pidemmän aikavälin tavoite. Solut oppivat käyttämään vapaita rasvahappoja energiasubstraatteina β-oksidaatiossa joitain viikkoja ketoosin alkamisen jälkeen. Aikaikkuna on varsin lavea, koska toisilla primaaristi β-oksidaatioon perustuva energia-aineenvaihdunta käynnistyy nopeammin kuin toisilla.
Tunnusomaista β-oksidaatioon siirtyneessä metaboliassa on ketoaineiden tuotannon väheneminen. Ruokailujen välillä rasvahappoja vapautuu tasaisena virtana rasvasoluista verenkiertoon, jossa ne kulkeutuvat soluihin ja hapettuvat β-oksidaatiossa energiaksi, mikä ylläpitää energistä, aktiivista, hieman euforista ja kylläistä oloa. Sama tapahtuu paastotessa.
Ketogeenisessä ruokavaliossa voi ja saa tehdä syrjähyppyjä. Jos mielesi tekee juoda lava bisseä, syödä perhepizza tai suklaalevy, anna palaa! Syrjähyppy ei ole maailmanloppu. Ketogeenisen ruokavalion tarkoituksena ei suinkaan ole kurjistaa elämää, vaan parantaa terveyttä ja elämänlaatua. Syrjähyppy on toki horjahdus ja askel taaksepäin, mutta se korjaantuu, kun ketogeenistä ruokavaliota jatkaa. Alussa syrjähyppyjen teko on helppoa ja houkuttelevaa, mutta pidempään ketoillessa syrjähypyn jälkeen on aivan yhtä helppoa ja luontevaa palata hiilihydraatteja rajoittavaan ruokavalioon. Ilmiö rinnastuu mielestäni alkoholin käyttöön: ihminen voi ja saa juoda toisinaan, mutta dokaamisesta ei pitäisi tehdä elämäntapaa. Ketogeeninen ruokavalio on elämäntapa, ei laihdutusruokavalio ja siksi minäkin vältän dieetistä puhumista. sanana dieetti rinnastuu vahvasti laihduttamiseen.
Yleensä kahden-kolmen päivän ketoilun jälkeen hiilihydraattien rajoittaminen johtaa siihen, että aivot alkavat käyttää solujen energiasubstraatteina enimmäkseen ketoaineita. Tämä aikaikkuna johtuu siitä, että maksan sokerivarastoissa, eli glykogeeneissä on glukoosia ihmisestä riippuen 1-3 päiväksi (~250 g) ja ketogeneesi käynnistyy glukoosivarastojen tyhjennyttyä.
96 tunnin kuluessa hiilihydraattien rajoittamisesta keskushermoston solut tyydyttävät suurimman osan ATP-tarpeestaan ketoaineilla [64]. Itse asiassa ketonit voivat toimia ATP-substraatteina ja tuottaa jopa 70% aivojen energiasta energiakysynnän tyydyttämiseksi [65, 66].
Alzheimerin taudin, dementian ja Parkinsonin taudin hoidossa kohonnut seerumin ketonipitoisuus (ketoosi) on lupaava terapiavaihtoehto [67–69]. Magneettikuvissa ketoosin on huomattu aktivoivan taudin passivoimia aivoalueita Alzheimerin tautia sairastavilla. Hyviä tuloksia on saatu myös dementiaa sairastavien potilaiden kognitiivisissa testeissä, joiden tulokset ovat glukoosin vähentymisen ja ketoaineiden lisääntyneen pitoisuuden seurauksena selvästi parantuneet.
Tähän on looginen selitys: hermosolujen glukoosinoton heikentyminen on yksi monista neurodegeneratiivisten tautien solutason vaurioitumisen syistä. Glukoosimetabolian heikentyesssä solut surkastuvat ja kuolevat energianpuutteeseen, mikä lisää aivojen atrofiaa ja ko. tautien oireita. Nämä solut kuitenkin saavat energiaa β-hydroksibutyraatista. Tämä ehkäisee solujen surkastumista ja dementian oireiden pahenemista. Taustalla oleva mekanismi on kiehtova.

4. Eksogeenisten ketonien käyttö
Endogeenisten ketonien muodostuminen on kehon normaali ja terveellinen selviytymismekanismi, jonka ansiosta ihminen selviää pitkään ilman ravintoa [58]. Tämä on ollut erityisen tärkeää esihistoriallisille esivanhemmillemme, joille ravinnon saanti päivittäin tai edes joka viikko ei ollut mikään itsestäänselvyys. Suuri muutos ravinnonsaannissa tapahtui oikestaan vasta maanviljelyn kehityttyä noin 10 000 vuotta sitten, jolloin ravintoa tuotettiin ja varastoitiin yli välittömän kulutuksen.
Metsästäjä-keräilijät elivät sillä, mitä löysivät tai saivat saaliiksi. Ruokaa syötiin silloin kun sitä oli. Ravinnosta saatu ylimääräinen energia varastoitiin rasvakudokseen. Aikoina, jolloin ravinnosta oli pulaa, solut tuottivat energiaa varastorasvasta. Ketogeneesi, glukoneogeneesi, rasvan β-oksidaatio ja perusaineenvaihdunnan hidastuminen pitävät ihmiset hengissä tarvittaessa useita viikkoja ilman ravintoa. Lihomisella on tärkeä fysiologinen tehtävä ihmisen selviytymisessä.
Solusignalointi
ATP-substraattina toimimisen lisäksi ketonit toimivat myös ligandeina, jotka säätelevät solujen signalointia ja käyttäytymistä [27]. Nämä edut toteutuvat vain, jos henkilö noudattaa ketogeenistä ruokavaliota. Ketogeenisen ruokavalion täydentäminen eksogeenisilla ketoaineilla voi ylläpitää ketoosiin perustuvaa aineenvaihduntaa pienistä syrjähypyistä huolimatta. Samanaikaisesti eksogeeniset ketonit edistävät suotuisaa farmakologiaa.
Ketonien tai proketonien (BHB) eksogeeninen käyttö lisäravinteena on ollut käytössä vuodesta 1975 alkaen. BHB (β-hydroksibutyraatti) muuttuu tarpeen mukaan muiksi ketoaineiksi, kuten asetoasetaatiksi tai alavirtaan asetoniksi. Asetoni ja asetoasetaatti ovat biologisia ketoneja, joista seerumin ketonipitoisuus suurimmaksi osaksi muodostuu [70].
Ketogeeninen ruokavalio ruokavalioterapiana aiheuttaa haasteita, koska se vaatii ylimääräistä omistautumista ja rruokavaliorajoituksia. LCHF voi johtaa siirtymäaikana huonovointisuutta aiheuttavaan ketoflunssaan. Joillekin ketoosin saavuttaminen on vaikeampaa kuin toisille metabolisten, geneettisten, ympäristön, sosiaalisten, kulttuuristen ja elämäntapoihin liittyvien tekijöiden vuoksi.
Eksogeeninen ketonilähde voi toimia siltana, joka kompensoi metaboliseen siirtymään liittyvää energiapuutetta, samalla kun se tarjoaa ketonilähteen, joka toimii solujen signalointiligandina. Sillä voi kuitenkin olla myös ruokavaliosta riippumaton rooli solunsisäisten signalointiominaisuuksiensa vuoksi.
Nykyisissä kaupallisissa ketoaineissa käytettyä suurta annostusta voidaan pitää tarpeettomana. Kuluttajille tarjotaan jopa 10 gramman BHB:tä yhdessä keskipitkäketjuisten triglyseridien (MCT) kanssa.
MCT toimii substraatina β-hapetukselle ja BHB:n muodostumiselle. Suun kautta otettava MCT liittyy monilla käyttäjillä ruoansulatuskanavan häiriöihin, kuten ripuliin [71–75]. Lisäksi nämä BHB-lisäravinteet sisältävät natriumia, jota voi olla 1300 mg annosta kohti. Terveydenhuollon ammattilaisen tulisi valvoa tällaisten erittäin suurten terapeuttisten annosten annostelua potilaille. Lisäravinteisiin liittyy aina yliannostuksen riski.

5. Eksogeenisten ketoaineiden hyödyt
Eksogeenisillä ketoaineilla, kuten BHB:llä (β-hydroksibutyraatilla) on on terapeuttista arvoa useiden sairauksien hoidossa. β-hydroksibutyraattilisän (BHB) in vivo -tutkimus vähensi syöpäkasvaimen kasvua ja pidensi tutkittavan kohteen eloonjäämistä muista ruokavalion tekijöistä, kuten seerumin glukoosipitoisuudesta riippumatta [76].
BHB:llä on havaittu tulehdusta hillitsevä vaikutus NLRP3-tulehduksen aiheuttaman IL-1β:n ja IL-18:n välittämisessä ihmisen monosyyteissä [77]. Tällä voi olla merkitystä autoinflammatoristen sairauksien hoidossa. Eksogeenisen ketonin tukema terapeuttinen ketoosi hillitsee epileptisten kohtausten alkamista [78].
β-hydroksibutyraatti (BHB) auttaa myös parantamaan sydämen terveyttä vähentämällä sydänlihaksen glukoosinottoa ja lisäämällä verenkiertoa [79]. Aivojen hypometabolisten sairauksien, kuten Alzheimerin taudin (AD), hoidossa käytetään menestyksekkäästi 10–20 grammaa eksogeenistä ketonilisää annoksiin jaettuna [80].
Alzheimerin tauti liittyy keskushermoston neuronien heikentyneeseen glukoosimetaboliaan, joka korreloi kognitiivisten kykyjen heikentymisen kanssa [81–84]. Ketoni ei ole riippuvainen insuliinista ja sitä voidaan käyttää hermosolujen mitokondrioissa tehokkaasti. β-hydroksibutyraatin saatavuus ehkäisee neurodegeneratiivisten tautien aivosolujen energiavajeen aiheuttamia solutuhoja [85].
Seerumitasojen ei tarvitse nousta merkittävästi, jotta aivojen energiansaantia voidaan tehostaa vaihtoehtoisella energiasubstraatilla. Tämä vähentää sivuoireiden riskiä ja minimoi hoidossa tarvittavan eksogeenisen ketoniannoksen.
Hyperketonemian, jossa systeemiset plasman ketonit nousevat vain tavallisten (0,2 mmol/l) perustasojen yli, on osoitettu parantavan aivojen ketonipitoisuutta ja tarjoavan neuroneille vaihtoehtoisen ja tehokkaan energiasubstraatin [80].
β-hydroksibutyraatti tukee mitokondrioiden energiantuotannon aktiivisuutta ja estää apoptoottisten (solukuolemaan indusoivien) proteiinien kumuloitumista neuroneihin [65]. Myrkytystilasta, vammoista tai iskemiasta johtuva neurodegeneraatio johtaa oksidatiiviseen stressiin. Eksogeenisten ketonien antaminen hiirimalleissa estää turvallisesti reaktiivisten happiradikaalien (ROS) muodostumista [86].
Ketogeenisen ruokavalion on dokumentoitu olevan tehokas hoito epilepsian ja lääkeresistentin epilepsian hoidossa [87, 88]. Eksogeenisten ketonien antamista on vuosikymmenien ajan käytetty hyvällä menestyksellä epilepsian hoitoon [78, 89].
Kokeellisessa rottamallissa eksogeenisten ketonien on havaittu lisäävän sekä rotan fyysistä aktiivisuutta että kognitiivista suorituskykyä [90]. Siitä, missä määrin eksogeeniset ketonit voivat säätää tai parantaa pitkittynyttä suorituskykyä ihmisillä, ei ole vielä tutkittua tietoa [91], mutta professori Tim Noakesin juoksemat ultramaratoonit ja triathlonisti Sami Inkisen käsittämättömät suoritukset ketogeenisellä ruokavaliolla viittaavat siihen, että ketogeeninen ruokavalio parantaa myös ihmisten henkistä ja fyysistä suorituskykyä.
Viime kädessä ketogeenisen ruokavalion vaikutuksia motivaatioon ja jaksamiseen tukee myös se, että minä multippelisklerootikkona käänsin, editoin ja uudelleenkirjoitin marraskuussa kahdeksan 10-25 A4-sivun mittaista tutkimuskatsausta Ruokasotaan. Kyllä sekin jotain kertoo ketogeenisestä ruokavaliosta ja sen vaikutuksista jaksamiseen.
Eksogeeniset ketonit voivat toimia terveyttä edistävinä aineina, mutta kuten myöhemmin osoitetaan, BHB:n ja sen molekyylisesti analogisen lyhytketjuisen rasvahapon, voihapon (BA) yhdistelmä voi olla tehokkaampi ja sopivampi terapiavaihtoehto mm. näiden yhteiskäytön tuoman synergiahyödyn vuoksi.

6. Eksogeenisten ketonien turvallisuus elintarvikkeissa ja hoidoissa
Ruoka sisältää useita luonnollisia ketonilähteitä. Maitotuotteet ja erityisesti täysmaito ovat luonnollisen β-hydroksibutyraatin lähteitä [92, 93]. Yhdysvaltain FDA luokittelee β-hydroksibutyraatin eri muodot yleisesti turvallisiksi (GRAS).
Eksogeeniset ketonit (tai ketoaineet) ovat turvallisia, mutta kuinka paljon on liikaa?
Koehenkilöt testasivat eksogeenisen ketonimäärän 395 mg / kg ketoniesterinä saantia aterian yhteydessä tai ilman. Seerumin BHB-tasot mitattiin tunnin kuluttua lisäravinteen antamisesta. Seerumin BHB oli alhaisempi BHB:n aterian rinnnalla saaneilla koehenkilöillä verrattuna niihin, jotka saivat BHB:n ilman ruokaa (2,1 mM ± 0,2 mM vs. 3,1 mM ± 0,1 Mm). Nämä äärimmäiset BHB-annokset muuttuivat 31,6 grammaksi ketoniestereitä 80 kg painavalla henkilöllä. Annos siedettiin hyvin [94].
Toisessa ihmiskokeessa käytettiin suun kautta annettua annosta (R) -3-hydroksibutyyli (R) -3-hydroksibutyraattia, joka on BHB-molekyylin monoesteri, kvantifioituna 714 mg / kg. Nämä annokset muuttuivat 57,1 grammaksi ketoniestereitä 80 kg painavalla koehenkilöllä. Maksimiplasman ketonit saavutettiin 2 tunnissa (3,30 mmol/l BHB ja 1,19 mmol/l asetoasetaatti). Tätä suurta annosta annettiin viiden päivän ajan kolme kertaa päivässä, ja myös se siedettiin hyvin [95] ilman sivuvaikutuksia.
Tyypillinen 8 tunnin paasto tuottaa 0,5 mmol/l seerumin ketonipitoisuuden [95]. Seitsemän paastopäivän aikana veren kokonaisketonitasot voivat nousta 5–7 mmol/l tasolle [25, 95].
Toksisuustutkimus rotilla, jotka saivat ketoaineita 12 ja 15 g / kg, tukee myös β-hydroksibutyraatin annostelun turvallisuutta [96].
Suun kautta annettu natrium D, L-β-hydroksibutyraatti (1000 mg / kg päivässä) on annettu alle 2-vuotiaille lapsille, joilla on kardiomyopatia ja leukodystrofia asyyli-CoA-dehydrogenaasipuutoksesta. Viikon kuluessa hoidon aloittamisesta havaittiin lasten toipumista täydellisestä halvauksesta. Kahden vuoden jälkeen todettiin neurologisen toiminnan huomattavaa parantumista. Lapset kävelivät ja aivojen MRI-kuvat osoitti selkeää toipumista.
Kaksi muuta samaa tilaa sairastavaa lasta, jotka eivät reagoineet tyypilliseen hoitoon, paranivat progressiivisesti edellä kuvatulla hoidolla [97]. Pikkulasten hyperinsulinemisessa hypoglykemiassa kahta kuuden kuukauden ikäistä lasta hoidettiin ja seurattiin viiden ja seitsemän kuukauden ajan. Lapsille annettiin neljän ja kahdeksan gramman ketoniannoksia, ja ne siedettiin hyvin [60].
On kuitenkin huomattava, että tällainen äärimmäinen terapeuttinen annostelu vaatii lääketieteellistä seurantaa.

7. Butyraatin terveyshyödyt
Lyhytketjuiset rasvahapot, joita kutsutaan myös haihtuviksi (volatile) rasvahapoiksi, ovat tyypillisesti suolen mikrobiomin tuottamia. Näitä rasvahappoja ovat butyraatti, propionaatti ja asetaatti, jotka syntyvät suolen symbioottisten mikrobien ravintokuidun käymisen sivutuotteina [98].
Suolistomikrobien tiedetään edistävän terveyttä ja hyvinvointia, vaikka ne vaikuttavat tavoilla, jotka ylittävät monimutkaisuudessaan immuunijärjestelmän toiminnan.
Nykyään tiedetään, että kommensaalibakteerit (normaalimikrobiston mikrobit, josta ei koidu isännälle hyötyä eikä haittaa) osallistuvat vitamiinien [99] synteesiin, ja tuottavat tärkeän energialähteen lyhytketjuisten rasvahappojen muodossa [100].
Lyhytketjuiset rasvahapot kiertävät takaisin säätääkseen ja ylläpitääkseen terveellistä suolistomikrobipopulaatiota siivoamalla luminaalisen (onteloon liittyvän) ympäristön patogeeneistä tyhjäksi [101, 102].
Luminaalibutyraatti lisää suoliston mikrobiomin hyvinvointia. Patogeenisiin bakteereihin, kuten koli-bakteereihin (Escherichia coli), salmonellaan (Salmonella spp.) ja kampylobakteereihin (Campylobacter spp.) luminaalibutyraatilla on negatiivinen vaikutus [103].
Butyraatin vaikutus ulottuu kuitenkin paksusuolen ulkopuolelle, jossa sitä syntyy. Butyraatti parantaa insuliiniherkkyyttä systeemisesti [102].
Suun kautta nautitun butyraatin on osoitettu indusoivan GLP-1:n eritystä [104]. Tämän hormonin tiedetään tukevan glukoositoleranssin ja ruokahalun hallintaa. Aivoissa GLP-1 tuottaa syvällisiä vaikutuksia, joiden mekanismit eivät ole aina selkeitä. Sen on osoitettu stimuloivan iskeemisten, eli paikalliseen verenpuutteeseen liittyvien vaurioiden neurogeneesiä aivopohjaisen neurotrofisen tekijän (BDNF) ylisääntelyn kautta [105]. Sillä on masennuslääkkeiden kaltaisia vaikutuksia [106].
Tutkimukset osoittavat, että butyraattia saaneet hiiret pysyvät hoikkina (ruokavalion kalorimäärästä huolimatta) [107]. Butyraatti on lisännyt hiirten energiankulutusta kehon lämmöntuotannon muodossa ja tehnyt hiiristä yleensä fyysisesti aktiivisempia [108, 109].
Butyraatilla on osoitettu olevan merkittävä sydän- ja verisuonitauteja ennaltaehkäisevä vaikutus [110, 111]. Tutkimuksissa butyraatti vähensi seerumin triglyseridejä peräti 50% verrokkeihin nähden [112]. Se myös vähentää endogeenisen kolesterolin tuotantoa [112].
Butyraatin ja asetaatin on todettu suojaavan ruokavalion aiheuttamalta lihavuudelta [107, 113]. Butyraatin antamisen on havaittu parantavan ruokahalua ja ravinteiden aineenvaihduntaa [114]. Butyraatti on avainpolttoaine suoliston epiteelisoluille ja se parantaa suolinukan eheyttä [115].
Aivan kuten BHB, butyraatti on histonideasetylaasien (HDAC) estäjä (inhibiittori), joka säätelee oksidatiivisen stressin vastustuskykyä koodaavien geenien transkriptiota [116].
HDAC-modulointi liittyy myös pitkäkestoiseen muistiin, oppimiseen ja neuronien välisten synaptisten yhteyksien plastisuuteen (neuroplastisuuteen) [117]. Aihe, johon täytyy pikimmiten tutustua!
Geenitranskription säätely johtaa myös parempaan suojaan vapailta happiradikaaleilta ja oksidatiivisen stressin aiheuttamilta kudosvaurioilta, joita voivat aiheuttaa äärimmäinen metabolinen stressi ja ympäristömyrkyt.

Butyraatin geenisäätely vaikuttaa neuroprotektiivisesti (aivosoluja suojaten) ja parantaen siten muistia esimerkiksi dementiassa [118]. Butyraatti estää NF-kB:tä ja lisääntyneitä I-kB-tasoja ja parantaa pitkäaikaista tulehduksen hallintaa [119].
Oraalisesti annettu natriumbutyraatti heikentää kokeellisesti indusoitua koliittia [120]. Suun kautta annetulla butyraatilla on myös tulehduksia estävä anti-inflammatorinen vaikutus. Se voi johtaa Crohnin taudin remissioon vähentämällä NF-kB: n ja IL-1β: n tasoa [121].
Suonensisäisesti annetun butyraatin on osoitettu tukevan suoraan ruoansulatuskanavan vuorauksen ja suolinukan terveyttä [103]. Sillä on vaikutuksia suoliston solujen lisääntymiseen ja solujen troofiseen ravinnonottoon 122].
Butyraatti on voimakas suoliston immuunipuolustusta säätelevien T-solujen promoottori [123]. Se luo immuunijärjestelmää säätelevän mekanismin, joka edistää parempaa tulehduksen hallintaa limakalvon vuorauksessa ja suolinukassa, sekä mekanismin suolistosyövän estämiseksi [124].
Butyraatti vähentää tai estää mikrobiomipopulaatiota, joka tuottaa propionihappoa [125]. Propionihappo on osallisena autismikirjon häiriöissä (ASD) [126]. On spekuloitu, että voihapon propionihappoa tuottavien suolistobakteerien säätelyvaikutus on mekanismi kognitiivisen tilan parantamiseksi [127].
70% lapsista, joilla on autismi tai ASD, on ruoansulatuskanavan häiriöitä ja muuttunut geenien ilmentyminen aivoissa. Sen on arveltu johtuvan lyhytketjuisten rasvahappojen epätasapainosta [128]. Butyraatin ja muiden lyhytketjuisten rasvahappojen oraalisten antoon liittyvien terveysetujen luettelo on pitkä (taulukko 1). β-hydroksibutyraatin antamisen yhteydessä butyraatti-lisä on suositeltava näiden yhteisvaikutusten vuoksi.
Veden passiivinen imeytyminen paksusuolessa riippuu lyhytketjuisten rasvahappojen saatavuudesta [129–131]. Butyraatilla on rooli terveessä peristaltiikassa, joka auttaa normalisoimaan suolessa liikkuvan massan liikettä ummetuksessa tai ripulissa [132, 133]. Butyraatti tukee optimaalista nesteytystä ja optimaalista suolen eliminointitoimintoa [134].
Tämä farmakologinen vaikutus auttaa torjumaan BHB-lisäravinteisiin liittyviä mahdollisia haittatapahtumia.
Yhteenveto butyraatin terveydellisistä hyödyistä, joita on raportoitu in vitro– ja in vivo -malleilla sekä ihmiskokeilla tehdyissä tutkimuksissa
Butyraattia saa runsaasti meijerituotteista. Voi, joka sisältää luonnostaan 3-4 % voihappoa, on itse asiassa yksi parhaimmista voihapon lähteistä. Yksi ruokalusikallinen voita (~14 g) sisältää ~560 mg voihappoa. Butyraatit ovat voihapon suoloja ja estereitä. Suolistossa esiintyvä voihappo näyttää hillitsevän tulehdusta ja syöpäsolujen kasvua sekä vähentävän happiradikaalien syntyä. Ihminen kuluttaa päivässä yli 1000 mg butyraattia ulkoisista lähteistä. Tämä saadaan ruokavalion rasvoista.
Ihmisillä, jotka noudattavat ketogeenistä ja / tai kaloreita rajoittavaa ruokavaliota, mutta eivät syö meijerituotteita (voita, kermaa ja juustoja), ja joiden kuitujen saanti ravinnosta on vähäistä, voihapon saanti ja synteesi suolistossa on kehon tarpeisiin nähden liian vähäistä. Butyraatin ottaminen lisäravinteena on perusteltua myös, koska se yhdistää ketogeenisen ruokavalion ja butyraattilisän edut synergisesti.
Butyraatti lisää FGF21:n pitoisuutta seerumissa, maksassa ja rasvasoluissa, mikä puolestaan stimuloi rasvahappojen β-hapettumista ja maksan ketonituotantoa [135, 136]. Tämä on butyraattifarmakologian keskeinen piirre, joka synergisoi suoraan sen aktiivisuuden ketogeeniseen aineenvaihduntaan ja tukee sen terveydellisiä vaikutuksia. Butyraatti itsessään voi myös toimia substraattina β-hapettumiselle [137].

8. Butyraatin (lyhytketjuisen rasvahapon) ja BHB: n yhdistämisen edut
Butyraatti toimii merkittävänä ketoosin induktiota kiihdyttävänä synergistisenä tekijänä, joka parantaa:
-
BHB-ligandivuorovaikutuksia ja farmakologiaa
-
yleistä terveydentilaa
-
kuntoa ja suorituskykyä
Ketonien, kuten BHB-suolan eksogeeninen saanti lisäravinteena tarjoaa aivosolujen ATP-tuotannolle välittömän vaihtoehtoisen energiasubstraatin kalori- tai hiilihydraattirajoituksen aikana.
Samanaikainen butyraattilisäys natriumin, kalsiumin tai kaliumbutyraatin (tai sen estereiden) muodossa:
-
indusoi elimistön endogeeniseen ketonisynteesin
-
toimii ligandina stimuloimalla reseptoreita, joihin ketonit vaikuttavat
-
myötävaikuttaa insuliinin ja aineenvaihdunnan yleisen terveyden parantamiseen
-
tukee tulehduksellista ja yleistä immuunijärjestelmän terveyttä
-
tukee neurologista terveyttä
-
tukee ruoansulatuskanavan terveyttä ja eheyttä
-
toimii suoraan ATP:n muodostamisen energiasubstraattina
Kaikki nämä toteutuvat rinnakkain niiden etujen kanssa, joita sisarketoaineen (BHB) samanaikainen lisäys tuottaa. Tämän synergistisen järjestelmän arvo ketogeenisen ruokavalion yhteydessä on hyvin perusteltu ja järkevä.
On kuitenkin muistettava, että ketogeeniselle elämäntavalle on ominaista vähäinen hiilihydraattien saanti, mikä johtaa heikentyneeseen sulamattoman kuidun ja resistentin tärkkelyksen saantiin. Sillä on negatiivinen vaikutus suoliston mikrobiomiin ja sen kykyyn tuottaa lyhytketjuisia rasvahappoja, kuten voihappoa.
Suoliston mikrobiomi on säännöllisesti kovan paineen alla ympäristötekijöiden, kuten ruokavalion ja lääkkeiden (esim. antibioottien) vaikutuksesta [138, 139]. Butyraatin ottaminen lisäravinteena suojaa suoliston mikrobiomia, etenkin jos sulamattomien kuitujen ja resistentin tärkkelyksen saanti on vähäistä.

9. Voihappo ja ketogeeninen painonpudotusstrategiaa
Lisäravinteena otetun BHB:n vaikutusta painonpudotuksessa on tutkittu hyvin paljon. Erityisen paljon huomiota on kiinnitetty lisäravinteisiin, jotka sisältävät BHB:n lisäksi keskipitkäketjuisia triglyseridejä (MCT). Ketoaineet ja MCT sisältävät energiaa ja lisäävät siten päivittäistä energiansaantia.
Tutkimuksissa on havaittu, että seerumin ketonipitoisuuden kasvu ei lisää, vaan estää lipolyysiä. Siltä kannalta lisäravinteena otetut ketoaineet ja MCT itse asiassa estävät rasvasolujen purkamista vapaiksi rasvahapoiksi, ketonien synteesiä ja laihtumista [53, 140]. Toisaalta butyraatti tukee ruokahalun hallintaa ja parantaa kehon rasva-lihas-koostumusta [107, 112–114].
On olemassa näyttöä, jonka mukaan butyraatti vaikuttaa suotuisasti sydän- ja verisuoniterveyteen ja ehkäisee sydän- ja verisuonitauteja [112]. Tasapaino eksogeenisten ja endogeenisten ketoaineiden välillä on oleellista aivojen ja kognitiivisen terveyden silloitustekijänä ja neuroniin liittyvien signaaliligandien riittävän saannin kannalta. Aktivointisignaali, kuten voihaposta peräisin oleva signaali rasvahappojen β-oksidaation käynnistämiseksi aivosoluissa, on neuronien toiminnan kannalta tärkeää.
Lisäravinteena otetutun butyraatin ja beta-hydroksibutyraatin käyttö on perusteltua ruokavalion siirtymäajalla sekä solujen energia-aineenvaihdunnan tehostajana monissa metabolisissa ja neurodegeneratiivisissa sairauksissa, mutta laihtumisen suhteen tällaisesta lisäravinnecocktailista ei ole hyötyä. Sen sijaan lisäaineina syiötävien butyraatin ja beta-hydroksibutyraatin hyödyntäminen paastolla tapahtuvan liikunnan energiabuusterina ja rasvahappojen hapettumisen tehostajana on perusteltua.
Ruokavalion tuottama ketoosi vähentää laktaatin tuotantoa ja parantaa suorituskykyä erityisesti kestävyyttä vaativissa lajeissa, kuten pyöräilyssä [141]. Sen on osoitettu estävän lihaskatoa (kataboliaa) ja suojaavan aivoja ja muita kudoksia hapettumiselta [142].

10. Kurkistus ketoaineiden solunsisäiseen farmakologiaan
BHB-BA-kompleksin farmakologiasta vastaavien mekanismien kartoittamiseksi ravintolisien yhteydessä on tehty useita tutkimuksia. Tutkimukset osoittavat, että erilaiset G-proteiiniin kytketyt HCA-reseptorit toimivat kohteina endogeenisille ketonille ja ketoaineiden ligandeille [143].
Tämä reseptoriperhe luokitellaan useisiin alatyyppeihin, joilla on erillisiä piirteitä, kuten ligandispesifisyys. Vaikka BHB toimii tehokkaana agonistina esimerkiksi HCA2-reseptoreille, se ei kykene toimimaan agonistina muille HCA-reseptoreille. Sekä BA että BHB ovat signalointiligandeja erilaisille reseptoreille, jotka osallistuvat neuroinflammatoriseen säätelyyn, mukaan lukien HCA2-reseptori [144].
Muut ligandit, kuten muut ketonit, voivat toimia agonisteina vaihtamalla HCA-reseptoreita, mutta ne eivät välttämättä pysty käynnistämään HCA2 –reseptorista transduktiokaskadia. HCA-reseptoreita voi esiintyä erilaisissa kudos- ja solutyypeissä, kuten rasvasoluissa ja makrofageissa [143].
Näiden reseptorien ilmentyminen voidaan myös indusoida immuunisoluissa, kuten makrofageissa, erilaisilla sytokiineilla ligandiensa solunsisäisen vaikutuksen säätelemiseksi. Vapaat rasvahappo- (FFAR) ja HCA-reseptorit voivat hyvinkin olla keskeisiä kohteita tyypin 2 diabeteksen, lihavuuden ja inflammaation ehkäisyssä ja hoidossa [145].
Ravinnetasapainoa ylläpitävät rasvahapporeseptorit, jotka säätelevät kolekystokiniiniä, peptidiä YY ja leptiiniä ovat kasvavan kiinnostuksen kohteena diabeteksen hoidossa.
Luonnossa esiintyvät ligandit, BHB ja BA moduloivat jo tehokkaasti näitä terapeuttisia kohteita. Kaikki kolme HCA-reseptoria ekspressoidaan rasvasoluissa. HCA1-reseptori aktivoidaan esimerkiksi hydroksipropaanihapolla (laktaatilla), kun taas HCA2:n agonisti on β-hydroksibutyraatti (BHB), ja HCA3 aktivoidaan toisella β-hapetusvälituotteella [146].
Näiden kahden luonnollisen butyraatin säätelyvaikutukset tulehduksellista kaskadia ja immuunijärjestelmän aktiivisuutta säätelevien sytokiinien transkriptiotekijöihin liittyvät läheisesti NF-kB-modulointiin.
Tumatekijä erytroidiin 2 liittyvä tekijä 2 (Nrf2) on ensisijainen transkriptiotekijä, joka käynnistää vasteen oksidatiiviseen stressiin. Ketogeeninen ruokavalio indusoi systemaattisesti Nrf2:ta lievän oksidatiivisen ja elektrofiilisen stressin kautta [147, 148].
Nrf2:n transkriptio avaa sarjan endogeenisiä antioksidanttisia puolustusjärjestelmiä. Transkriptiotekijä siirtyy tumaan ja sitoo antioksidanttivaste-elementin (ARE) transkriptoimaan solua suojaavat sytoprotektiiviset geenit [149].
Nrf2 transkriptoi endogeeniset antioksidanttipeptidit: hemeoksigenaasi-1, katalaasi (CAT), superoksididismutaasi (SOD) ja glutationiperoksidaasi (GSH / GPx) [150-152] oksidatiivisen stressin suojamekanismina. Viime aikoina tätä mekanismia on kohdennettu kemopreventiivisesti, millä on haluttu stimuloida endogeenista antioksidanttisaturaatiota, joka estää syöpä- ja kemoterapialääkkeiden aiheuttamat vahingot isäntäsolun terveessä DNA:ssa [153, 154].
Nrf2 lisää solujen puolustusmekanismeja. Se välittää mitokondrioille hermosuojauksen toksiinin aiheuttaman stressin aikana ja ehkäisee vaurioiden (leesioiden) muodostumista [155, 156].
Tätä solusuojausta nähdään myös kemoterapian yhteydessä, jossa Nrf2-induktio suojaa terveitä soluja [157]. Nrf2-induktio suojaa soluja LPS:n aiheuttamalta tulehdukselliselta aktiivisuudelta ja kuolleisuudelta [158].
Nrf2-signalointireitit ovat lupaavia Parkinsonin taudin mitokondrioiden toimintahäiriöiden vastatoimena [159]. Nrf2-induktion välittää myös puolustuksen sydänlihassolujen kohonneesta seerumin-glukoosin aiheuttamasta oksidatiivisesta vahingosta [160].
Diabeettinen tila liittyy Nrf2-aktiivisuuden alasregulaatioon ERK:n kautta. Tämän uskotaan vaikuttavan stressin aiheuttamaan insuliiniresistenssiin sydämen soluissa [161]. Tutkimukset osoittavat, että Nrf2-aktivaatiota voidaan käyttää terapeuttisena sovelluksena diabeteksen ”metabolisen häiriön parantamiseen ja munuaisvaurioiden lievittämiseen” [162].
Nrf2:n rooli solujen suojauksessa antioksidanttisen puolustuksen pääregulaattorina tekevät siitä kiinnostavan kohteen kudosten ja solujen suojaamisessa hapettavilta ja toksisilta tekijöiltä [163, 164]. Nrf2:lla on huomattava merkitys antioksidanttipuolustusmekanismissa muiden yleisten endogeenisten antioksidanttien rinnalla. Se tukee myös vammoista, toksisuudesta ja hypoksiasta palautumista [165, 166].
Iskemia (paikallinen hapenpuute) on yleinen solun toimintahäiriön ja solukuoleman syy. Iskemia johtuuu verenkierron keskeytymisestä tai hapen saatavuuden heikkenemisestä kudoksissa, mikä johtaa soluvaurioihin. Sen tiedetään olevan keskeinen tekijä aivohalvauksen patologiassa ja yksi yleisimmistä pysyvien solu- ja kudosvaurioiden aiheuttajista sydänsairauksissa [167].
Hemeoksigenaasi-1-induktio suojaa neuroneja [168] ja sydänkudosta [169] iskemialta ja sen aiheuttamilta vaurioilta. Myös glutationiperoksidaasin yliekspressio suojaa sydänlihasta iskeemisiltä reperfuusiovaurioilta [170, 171].
Butyraatti aktivoi Nrf2:ta [172, 173]. Tutkimuskirjallisuudessa on viitteitä siitä, että käsittely butyraatilla tai sen suoloilla (natriumbutyraatilla) lievittää oksidatiivista stressiä [174] ja parantaa katalaasiaktiivisuutta [175]. Esikäsittely BA-annoksella suojaa iskemiaan liittyviä sydänlihaksen vaurioita estämällä tulehduksellisten sytokiinien ilmentymistä [174].
Se myös suojaa keuhkovaltimon sileän lihaksen soluja hyperoksiaan liittyvältä hapettumiselta [175] ja parantaa ikääntymiseen liittyvää aineenvaihduntaa ja lihasten surkastumista [176].

11. Opittavaa on paljon
Monet voivat hyötyä ketogeenisestä ruokavaliosta tai suun kautta otettavista ketoaineista ja niiden tuottamasta ketoositilasta.
Ketoosi ylläpitää parempaa ruokahalun hallintaa, fyysistä kuntoa, aivojen tehostunutta energiansaantia, neuroplastisuutta, neurogeneesiä, oppimiskykyä ja parempaa muistia. Ketoosin aiheuttama beta-oksidaatio ylläpitää tasaista eenergiavirtaa, joka lisää kestävyyttä ja polttaa tehokkaasti rasvaa.
Solutasolla ketonit vaikuttavat neuro- ja sytoprotektiivisesti suojaten soluja ja hillitsevät vapaiden happiradikaalien ja oksidatiivisen stressin aiheuttamia solu- ja kudosvaurioita. Tutkimuskirjallisuuden meta-analyysin perusteella ketoosin hyötyjä ovat:
-
tulehduksen (inflammaation) lievittäminen
-
neurologiseen sairauteen liittyvä kognitiivisen heikentymisen korjaantuminen
-
parantunut ruoansulatuskanavan terveys
-
nopeampi palautuminen liikunnan tai intensiivisen harjoituksen lihasrasituksesta
Lisää työtä ja kliinisiä tutkimuksia tarvitaan, jotta tiedämme tarkemmin, miten näitä strategioita voidaan käyttää potilaiden terapiana.
12. Keskustelua
Tutkimuskirjallisuuden tämänhetkisen näytön perusteella lisäravinteena otetun eksogeenisen ketonin käyttö näyttää olevan toteuttamiskelpoinen strategia, joka tukee ketogeenisen ruokavalion siirtymävaihetta, jossa keho totutetaan glukoosin sijaan uuteen energiasubstraattiin. Butyraatinn on raportoitu antavan positiivisia tuloksia kunto-, painonhallinta-, kognitio- ja suorituskyvyn parantamisen tueksi joko ruokavalion rajoituksilla tai ilman.
Laboratoriomme nykyinen tutkimushanke on suunniteltu tutkimaan edelleen BHB:n ja BHB-BA:n solunsisäisiä vaikutuksia immuunijärjestelmän tärkeimpiin soluihin seerumipitoisuuksilla, jotka voimme saavuttaa suositellulla vähimmäisannoksella.
Eksogeeninen BHB-BA-ravintolisä voi olla toiminnallinen strategia, joka indusoi β-hapettumista ja auttaa nostamaan seerumin ketonitasoja, jotka tuottavat ketoosin (> 0,2 mmol) metaboliset hyödyt ilman makroravinteiden ankaraa säätelyä. BHB:n samanaikainen antaminen siihen liittyvän BA-molekyylin kanssa näyttää olevan tehokas tapa saavuttaa tämä tavoite käyttämällä erittäin pieniä ja turvallisia oraalisia annoksia. Vaikka ketoosin metabolisia hyötyjä saatetaan saavuttaa lisäravinteilla, on todennäköistä, että ketogeeninen ruokavalio yhdessä lisäravinteina otettavien butyraatin ja beta-hydroksibutyraatin kanssa toimii terapiana etenkin kognitiivisten häiriöiden ja painonhallinnan yhteydessä paremmin kuin lisäravinteet yksin.
Ruokavalion täydentäminen BHB-BA-lisäravinteella tukee ketoosissa pysymistä pienistä ruokavaliolipsahduksista huolimatta.
Huomio: Ota yhteys lääkäriin ennenBHB-BA-lisäravinteiden käyttöä. Älä käytä, jos olet raskaana tai imetät. Ei suositella tyypin I diabeetikoille.
Ps. Pahoittelut kirjoitus- ja/tai asiavirheistä. Nppäilyvirheille tulee jotenkin sokeaksi.
Conflicts of InterestFranco Cavaleri is the owner of a biomedical research group, Biologic Nutrigenomics Health Research Corp., and Biologic Pharmamedical Research that funds and executes research on the pharmacology of nutritional, nutraceutical, and pharmaceutical agents that are studied in the context of disease pathology including characteristics that have been associated with inflammation and dementias. Franco Cavaleri is also the owner of ketone-based and other related intellectual properties. Emran Bashar is an employee of the Biologic Pharmamedical Research.Authors’ ContributionsFranco Cavaleri was responsible for background research and preparation and editing of the manuscript. Emran Bashar was responsible for conducting research and preparation and editing of the manuscript. Franco Cavaleri and Emran Bashar generated research plans. |
 Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/
Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/
References
- A. Gjedde and C. Crone, “Induction processes in blood-brain transfer of ketone bodies during starvation,” American Journal of Physiology–Legacy Content, vol. 229, no. 5, pp. 1165–1169, 1975. View at: Publisher Site | Google Scholar
- M. Pollay and F. Alan Stevens, “Starvation-induced changes in transport of ketone bodies across the blood-brain barrier,” Journal of Neuroscience Research, vol. 5, no. 2, pp. 163–172, 1980. View at: Publisher Site | Google Scholar
- S. Cunnane, S. Nugent, M. Roy et al., “Brain fuel metabolism, aging, and Alzheimer’s disease,” Nutrition, vol. 27, no. 1, pp. 3–20, 2011. View at: Publisher Site | Google Scholar
- M. A. Reger, S. T. Henderson, C. Hale et al., “Effects of β-hydroxybutyrate on cognition in memory-impaired adults,” Neurobiology of Aging, vol. 25, no. 3, pp. 311–314, 2004. View at: Publisher Site | Google Scholar
- L. C. Costantini, L. J. Barr, J. L. Vogel, and S. T. Henderson, “Hypometabolism as a therapeutic target in Alzheimer’s disease,” BMC Neuroscience, vol. 9, no. 2, p. S16, 2008. View at: Publisher Site | Google Scholar
- W. R. Leonard, “Dietary change was a driving force in human evolution,” Scientific American, vol. 287, no. 6, pp. 106–116, 2002. View at: Publisher Site | Google Scholar
- S. M. Innis, “Dietary (n−3) fatty acids and brain development,” Journal of Nutrition, vol. 137, no. 4, pp. 855–859, 2007. View at: Publisher Site | Google Scholar
- E. Cohen, M. Cragg, A. Hite, M. Rosenberg, and B. Zhou, “Statistical review of US macronutrient consumption data, 1965–2011: Americans have been following dietary guidelines, coincident with the rise in obesity,” Nutrition, vol. 31, no. 5, pp. 727–732, 2015. View at: Publisher Site | Google Scholar
- J. Scholl, “Traditional dietary recommendations for the prevention of cardiovascular disease: do they meet the needs of our patients?” Cholesterol, vol. 2012, pp. 1–9, 2012. View at: Publisher Site | Google Scholar
- G. Mullins, C. Hallam, and I. Broom, “Ketosis, ketoacidosis and very-low-calorie diets: putting the record straight,” Nutrition Bulletin, vol. 36, no. 3, pp. 397–402, 2011. View at: Publisher Site | Google Scholar
- D. K. Layman and D. A. Walker, “Potential importance of leucine in treatment of obesity and the metabolic syndrome,” Journal of Nutrition, vol. 136, no. 1, pp. 319S–323S, 2006. View at: Publisher Site | Google Scholar
- M. Lawson and V. Shaw, “Ketogenic diet for epilepsy,” in Clinical Paediatric Dietetics, pp. 222–232, Blackwell Science Ltd., Oxford, UK, 2nd edition, 2001. View at: Google Scholar
- R. Krikorian, M. D. Shidler, K. Dangelo, S. C. Couch, S. C. Benoit, and D. J. Clegg, “Dietary ketosis enhances memory in mild cognitive impairment,” Neurobiology of Aging, vol. 33, no. 2, pp. 425. e19–425. e27, 2012. View at: Publisher Site | Google Scholar
- K. W. Barañano and A. L. Hartman, “The ketogenic diet: uses in epilepsy and other neurologic illnesses,” Current Treatment Options in Neurology, vol. 10, no. 6, pp. 410–419, 2008. View at: Publisher Site | Google Scholar
- P. G. Sullivan, N. A. Rippy, K. Dorenbos, R. C. Concepcion, A. K. Agarwal, and J. M. Rho, “The ketogenic diet increases mitochondrial uncoupling protein levels and activity,” Annals of Neurology, vol. 55, no. 4, pp. 576–580, 2004. View at: Publisher Site | Google Scholar
- E. C. Westman, J. Mavropoulos, W. S. Yancy Jr., and J. S. Volek, “A review of low-carbohydrate ketogenic diets,” Current Atherosclerosis Reports, vol. 5, no. 6, pp. 476–483, 2003. View at: Publisher Site | Google Scholar
- K. M. Maruschak, Impact of a Low-Carbohydrate, High-Fat Modified Ketogenic Diet on Anthropometrics, Biochemical Values, and Gastrointestinal Symptoms in Adult Patients with Epilepsy, Rush University, Chicago, IL, USA, 2016.
- S. R. Send, The Impact of a Low-Carbohydrate, High-Fat Modified Ketogenic Diet on Seizure Severity, Seizure Frequency, and Quality of Life in Adult Patients with Epilepsy, Rush University, Chicago, IL, USA, 2016.
- C. Dudick, “Carb”(not “Keto”) is a Four Letter Word, 2016.
- M. Schmidt, N. Pfetzer, M. Schwab, I. Strauss, and U. Kämmerer, “Effects of a ketogenic diet on the quality of life in 16 patients with advanced cancer: a pilot trial,” Nutrition and Metabolism, vol. 8, no. 1, p. 54, 2011. View at: Publisher Site | Google Scholar
- D. K. Layman and J. I. Baum, “Dietary protein impact on glycemic control during weight loss,” Journal of Nutrition, vol. 134, no. 4, pp. 968S–973S, 2004. View at: Publisher Site | Google Scholar
- C. Remesy, P. Fafournoux, and C. Demigne, “Control of hepatic utilization of serine, glycine and threonine in fed and starved rats,” Journal of Nutrition, vol. 113, no. 1, pp. 28–39, 1983. View at: Publisher Site | Google Scholar
- N. J. Krilanovich, “Benefits of ketogenic diets,” American Journal of Clinical Nutrition, vol. 85, no. 1, pp. 238-239, 2007. View at: Publisher Site | Google Scholar
- D. W. Kim, H. C. Kang, J. C. Park, and H. D. Kim, “Benefits of the nonfasting ketogenic diet compared with the initial fasting ketogenic diet,” Pediatrics, vol. 114, no. 6, pp. 1627–1630, 2004. View at: Publisher Site | Google Scholar
- R. L. Veech, “The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism,” Prostaglandins, Leukotrienes and Essential Fatty Acids, vol. 70, no. 3, pp. 309–319, 2004. View at: Publisher Site | Google Scholar
- J. D. McGarry, “Disordered metabolism in diabetes: have we underemphasized the fat component?” Journal of Cellular Biochemistry, vol. 55, no. S1994A, pp. 29–38, 1994. View at: Publisher Site | Google Scholar
- J. C. Newman and E. Verdin, “Ketone bodies as signaling metabolites,” Trends in Endocrinology and Metabolism, vol. 25, no. 1, pp. 42–52, 2014. View at: Publisher Site | Google Scholar
- O. Owen, G. Reichard Jr., H. Markus, G. Boden, M. Mozzoli, and C. Shuman, “Rapid intravenous sodium acetoacetate infusion in man metabolic and kinetic responses,” Journal of Clinical Investigation, vol. 52, no. 10, pp. 2606–2616, 1973. View at: Publisher Site | Google Scholar
- E. O. Balasse and F. Féry, “Ketone body production and disposal: effects of fasting, diabetes, and exercise,” Diabetes/Metabolism Reviews, vol. 5, no. 3, pp. 247–270, 1989. View at: Publisher Site | Google Scholar
- R. Wilson and W. Reeves, “Neutrophil phagocytosis and killing in insulin-dependent diabetes,” Clinical and Experimental Immunology, vol. 63, no. 2, p. 478, 1986. View at: Google Scholar
- M. Brownlee, H. Vlassara, A. Kooney, P. Ulrich, and A. Cerami, “Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking,” Science, vol. 232, no. 4758, pp. 1629–1632, 1986. View at: Publisher Site | Google Scholar
- N. Ahmed, “Advanced glycation endproducts—role in pathology of diabetic complications,” Diabetes Research and Clinical Practice, vol. 67, no. 1, pp. 3–21, 2005. View at: Publisher Site | Google Scholar
- P. Marceau, S. Biron, F. S. Hould et al., “Liver pathology and the metabolic syndrome X in severe obesity,” Journal of Clinical Endocrinology and Metabolism, vol. 84, no. 5, pp. 1513–1517, 1999. View at: Publisher Site | Google Scholar
- M. Y. Donath and S. E. Shoelson, “Type 2 diabetes as an inflammatory disease,” Nature Reviews Immunology, vol. 11, no. 2, pp. 98–107, 2011. View at: Publisher Site | Google Scholar
- K. Moley, M. Y. Chi, C. Knudson, S. Korsmeyer, and M. Mueckler, “Hyperglycemia induces apoptosis in pre-implantation embryos through cell death effector pathways,” Nature Medicine, vol. 4, no. 12, pp. 1421–1424, 1998. View at: Publisher Site | Google Scholar
- S. P. Hays, E. B. Smith, and A. L. Sunehag, “Hyperglycemia is a risk factor for early death and morbidity in extremely low birth-weight infants,” Pediatrics, vol. 118, no. 5, pp. 1811–1818, 2006. View at: Publisher Site | Google Scholar
- H. Vlassara, “Advanced glycation end-products and atherosclerosis,” Annals of Medicine, vol. 28, no. 5, pp. 419–426, 1996. View at: Publisher Site | Google Scholar
- H. Yki-Jarvinen, “Glucose Toxicity∗,” Endocrine Reviews, vol. 13, no. 3, pp. 415–431, 1992. View at: Publisher Site | Google Scholar
- L. L. Madison, D. Mebane, R. H. Unger, and A. Lochner, “The hypoglycemic action of ketones. II. Evidence for a stimulatory feedback of ketones on the pancreatic beta cells,” Journal of Clinical Investigation, vol. 43, no. 3, pp. 408–415, 1964. View at: Publisher Site | Google Scholar
- A. Baron, G. Brechtel, and S. Edelman, “Effects of free fatty acids and ketone bodies on in vivo non-insulin-mediated glucose utilization and production in humans,” Metabolism, vol. 38, no. 11, pp. 1056–1061, 1989. View at: Publisher Site | Google Scholar
- T. A. Hussain, T. C. Mathew, A. A. Dashti, S. Asfar, N. Al-Zaid, and H. M. Dashti, “Effect of low-calorie versus low-carbohydrate ketogenic diet in type 2 diabetes,” Nutrition, vol. 28, no. 10, pp. 1016–1021, 2012. View at: Publisher Site | Google Scholar
- T. D. Noakes, “Low-carbohydrate and high-fat intake can manage obesity and associated conditions: occasional survey,” South African Medical Journal, vol. 103, no. 11, pp. 826–830, 2013. View at: Publisher Site | Google Scholar
- J. Ratliff, G. Mutungi, M. J. Puglisi, J. S. Volek, and M. L. Fernandez, “Carbohydrate restriction (with or without additional dietary cholesterol provided by eggs) reduces insulin resistance and plasma leptin without modifying appetite hormones in adult men,” Nutrition Research, vol. 29, no. 4, pp. 262–268, 2009. View at: Publisher Site | Google Scholar
- J. S. Volek, M. J. Sharman, D. M. Love, N. G. Avery, T. P. Scheett, and W. J. Kraemer, “Body composition and hormonal responses to a carbohydrate-restricted diet,” Metabolism, vol. 51, no. 7, pp. 864–870, 2002. View at: Publisher Site | Google Scholar
- C. A. Major, M. J. Henry, M. de Veciana, and M. A. Morgan, “The effects of carbohydrate restriction in patients with diet-controlled gestational diabetes,” Obstetrics and Gynecology, vol. 91, no. 4, pp. 600–604, 1998. View at: Publisher Site | Google Scholar
- A. Accurso, R. K. Bernstein, A. Dahlqvist et al., “Dietary carbohydrate restriction in type 2 diabetes mellitus and metabolic syndrome: time for a critical appraisal,” Nutrition and Metabolism, vol. 5, no. 1, p. 9, 2008. View at: Publisher Site | Google Scholar
- R. D. Feinman, W. K. Pogozelski, A. Astrup et al., “Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base,” Nutrition, vol. 31, no. 1, pp. 1–13, 2015. View at: Publisher Site | Google Scholar
- M. K. Badman, A. R. Kennedy, A. C. Adams, P. Pissios, and E. Maratos-Flier, “A very low carbohydrate ketogenic diet improves glucose tolerance in ob/ob mice independently of weight loss,” American Journal of Physiology-Endocrinology and Metabolism, vol. 297, no. 5, pp. E1197–E1204, 2009. View at: Publisher Site | Google Scholar
- K. Xu, X. Sun, B. O. Eroku, C. P. Tsipis, M. A. Puchowicz, and J. C. LaManna, “Diet-induced ketosis improves cognitive performance in aged rats,” in Advances in Experimental Medicine and Biology, pp. 71–75, Springer, Berlin, Germany, 2010. View at: Google Scholar
- K. D. Ballard, E. E. Quann, B. R. Kupchak et al., “Dietary carbohydrate restriction improves insulin sensitivity, blood pressure, microvascular function, and cellular adhesion markers in individuals taking statins,” Nutrition Research, vol. 33, no. 11, pp. 905–912, 2013. View at: Publisher Site | Google Scholar
- R. A. Hawkins, A. M. Mans, and D. W. Davis, “Regional ketone body utilization by rat brain in starvation and diabetes,” American Journal of Physiology-Endocrinology and Metabolism, vol. 250, no. 2, pp. E169–E178, 1986. View at: Publisher Site | Google Scholar
- T. N. Seyfried and P. Mukherjee, “Targeting energy metabolism in brain cancer: review and hypothesis,” Nutrition and Metabolism, vol. 2, no. 1, p. 30, 2005. View at: Publisher Site | Google Scholar
- L. Laffel, “Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes,” Diabetes/Metabolism Research and Reviews, vol. 15, no. 6, pp. 412–426, 1999. View at: Publisher Site | Google Scholar
- H. Krebs, “The regulation of the release of ketone bodies by the liver,” Advances in Enzyme Regulation, vol. 4, pp. 339–353, 1966. View at: Publisher Site | Google Scholar
- J. McGarry and D. Foster, “Regulation of hepatic fatty acid oxidation and ketone body production,” Annual Review of Biochemistry, vol. 49, no. 1, pp. 395–420, 1980. View at: Publisher Site | Google Scholar
- M. T. Newport, T. B. VanItallie, Y. Kashiwaya, M. T. King, and R. L. Veech, “A new way to produce hyperketonemia: use of ketone ester in a case of Alzheimer’s disease,” Alzheimer’s and Dementia, vol. 11, no. 1, pp. 99–103, 2015. View at: Publisher Site | Google Scholar
- E. C. Westman, R. D. Feinman, J. C. Mavropoulos et al., “Low-carbohydrate nutrition and metabolism,” American Journal of Clinical Nutrition, vol. 86, no. 2, pp. 276–284, 2007. View at: Publisher Site | Google Scholar
- C. Harvey, What is Nutritional Ketosis? 2015.
- I. F. Kodde, J. van der Stok, R. T. Smolenski, and J. W. de Jong, “Metabolic and genetic regulation of cardiac energy substrate preference,” Comparative Biochemistry and Physiology Part A: Molecular and Integrative Physiology, vol. 146, no. 1, pp. 26–39, 2007. View at: Publisher Site | Google Scholar
- B. Plecko, S. Stoeckler-Ipsiroglu, E. Schober et al., “Oral β-hydroxybutyrate supplementation in two patients with hyperinsulinemic hypoglycemia: monitoring of β-hydroxybutyrate levels in blood and cerebrospinal fluid, and in the brain by in vivo magnetic resonance spectroscopy,” Pediatric Research, vol. 52, no. 2, pp. 301–306, 2002. View at: Publisher Site | Google Scholar
- H. White and B. Venkatesh, “Clinical review: ketones and brain injury,” Critical Care, vol. 15, no. 2, p. 219, 2011. View at: Publisher Site | Google Scholar
- E. P. Vining, “Clinical efficacy of the ketogenic diet,” Epilepsy Research, vol. 37, no. 3, pp. 181–190, 1999. View at: Publisher Site | Google Scholar
- E. H. Kossoff, B. A. Zupec-Kania, and J. M. Rho, “Ketogenic diets: an update for child neurologists,” Journal of Child Neurology, vol. 24, no. 8, pp. 979–988, 2009. View at: Publisher Site | Google Scholar
- G. F. Cahill Jr., “Fuel metabolism in starvation,” Annual Review of Nutrition, vol. 26, no. 1, pp. 1–22, 2006. View at: Publisher Site | Google Scholar
- M. Gasior, M. A. Rogawski, and A. L. Hartman, “Neuroprotective and disease-modifying effects of the ketogenic diet,” Behavioural Pharmacology, vol. 17, no. 5-6, pp. 431–439, 2006. View at: Publisher Site | Google Scholar
- R. de Oliveira Caminhotto and F. B. Lima, “Low carbohydrate high fat diets: when models do not match reality,” Archives of Endocrinology and Metabolism, vol. 60, no. 4, pp. 405-406, 2016. View at: Publisher Site | Google Scholar
- M. G. Abdelwahab, S. H. Lee, D. O’Neill et al., “Ketones prevent oxidative impairment of hippocampal synaptic integrity through K ATP channels,” PLoS One, vol. 10, no. 4, Article ID e0119316, 2015. View at: Publisher Site | Google Scholar
- J. X. Yin, M. Maalouf, P. Han et al., “Ketones block amyloid entry and improve cognition in an Alzheimer’s model,” Neurobiology of Aging, vol. 39, pp. 25–37, 2016. View at: Publisher Site | Google Scholar
- J. Zhang, Q. Cao, S. Li et al., “3-Hydroxybutyrate methyl ester as a potential drug against Alzheimer’s disease via mitochondria protection mechanism,” Biomaterials, vol. 34, no. 30, pp. 7552–7562, 2013. View at: Publisher Site | Google Scholar
- L. Siegel, N. I. Robin, and L. J. McDonald, “New approach to determination of total ketone bodies in serum,” Clinical Chemistry, vol. 23, no. 1, pp. 46–49, 1977. View at: Google Scholar
- D. J. Angus, M. Hargreaves, J. Dancey, and M. A. Febbraio, “Effect of carbohydrate or carbohydrate plus medium-chain triglyceride ingestion on cycling time trial performance,” Journal of Applied Physiology, vol. 88, no. 1, pp. 113–119, 2000. View at: Publisher Site | Google Scholar
- L. Misell, N. Lagomarcino, V. Schuster, and M. Kern, “Chronic medium-chain triacylglycerol consumption and endurance performance in trained runners,” Journal of Sports Medicine and Physical Fitness, vol. 41, no. 2, p. 210, 2001. View at: Google Scholar
- V. Ööpik, S. Timpmann, L. Medijainen, and H. Lemberg, “Effects of daily medium-chain triglyceride ingestion on energy metabolism and endurance performance capacity in well-trained runners,” Nutrition Research, vol. 21, no. 8, pp. 1125–1135, 2001. View at: Publisher Site | Google Scholar
- Y. M. C. Liu, “Medium-chain triglyceride (MCT) ketogenic therapy,” Epilepsia, vol. 49, no. s8, pp. 33–36, 2008. View at: Publisher Site | Google Scholar
- A. E. Jeukendrup, W. Saris, P. Schrauwen, F. Brouns, and A. Wagenmakers, “Metabolic availability of medium-chain triglycerides coingested with carbohydrates during prolonged exercise,” Journal of Applied Physiology, vol. 79, no. 3, pp. 756–762, 1995. View at: Publisher Site | Google Scholar
- A. Poff, C. Ari, P. Arnold, T. Seyfried, and D. D’Agostino, “Ketone supplementation decreases tumor cell viability and prolongs survival of mice with metastatic cancer,” International Journal of Cancer, vol. 135, no. 7, pp. 1711–1720, 2014. View at: Publisher Site | Google Scholar
- Y. H. Youm, K. Y. Nguyen, R. W. Grant et al., “The ketone metabolite [beta]-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease,” Nature Medicine, vol. 21, no. 3, pp. 263–269, 2015. View at: Publisher Site | Google Scholar
- D. P. D’Agostino, R. Pilla, H. E. Held et al., “Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats,” American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, vol. 304, no. 10, pp. R829–R836, 2013. View at: Publisher Site | Google Scholar
- L. C. Gormsen, M. Svart, H. H. Thomsen et al., “Ketone body infusion with 3-hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: a positron emission tomography study,” Journal of the American Heart Association, vol. 6, no. 3, p. e005066, 2017. View at: Publisher Site | Google Scholar
- S. T. Henderson, J. L. Vogel, L. J. Barr, F. Garvin, J. J. Jones, and L. C. Costantini, “Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial,” Nutrition and Metabolism, vol. 6, no. 1, p. 31, 2009. View at: Publisher Site | Google Scholar
- E. Arnaiz, V. Jelic, O. Almkvist et al., “Impaired cerebral glucose metabolism and cognitive functioning predict deterioration in mild cognitive impairment,” Neuroreport, vol. 12, no. 4, pp. 851–855, 2001. View at: Publisher Site | Google Scholar
- C. X. Gong, F. Liu, and K. Iqbal, “Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation,” Journal of Alzheimer’s Disease, vol. 9, no. 1, pp. 1–12, 2006. View at: Publisher Site | Google Scholar
- C. Messier, “Impact of impaired glucose tolerance and type 2 diabetes on cognitive aging,” Neurobiology of Aging, vol. 26, no. 1, pp. 26–30, 2005. View at: Publisher Site | Google Scholar
- S. T. Henderson, “Ketone bodies as a therapeutic for Alzheimer’s disease,” Neurotherapeutics, vol. 5, no. 3, pp. 470–480, 2008. View at: Publisher Site | Google Scholar
- T. B. VanItallie and T. H. Nufert, “Ketones: metabolism’s ugly duckling,” Nutrition Reviews, vol. 61, no. 10, pp. 327–341, 2003. View at: Publisher Site | Google Scholar
- M. Maalouf, P. G. Sullivan, L. Davis, D. Y. Kim, and J. M. Rho, “Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation,” Neuroscience, vol. 145, no. 1, pp. 256–264, 2007. View at: Publisher Site | Google Scholar
- C. B. Henderson, F. M. Filloux, S. C. Alder, J. L. Lyon, and D. A. Caplin, “Efficacy of the ketogenic diet as a treatment option for epilepsy: meta-analysis,” Journal of Child Neurology, vol. 21, no. 3, pp. 193–198, 2006. View at: Publisher Site | Google Scholar
- J. Sirven, B. Whedon, D. Caplan et al., “The ketogenic diet for intractable epilepsy in adults: preliminary results,” Epilepsia, vol. 40, no. 12, pp. 1721–1726, 1999. View at: Publisher Site | Google Scholar
- M. A. McNally and A. L. Hartman, “Ketone bodies in epilepsy,” Journal of Neurochemistry, vol. 121, no. 1, pp. 28–35, 2012. View at: Publisher Site | Google Scholar
- A. J. Murray, N. S. Knight, M. A. Cole et al., “Novel ketone diet enhances physical and cognitive performance,” Federation of American Societies for Experimental Biology Journal, vol. 30, no. 12, pp. 4021–4032, 2016. View at: Publisher Site | Google Scholar
- P. J. Pinckaers, T. A. Churchward-Venne, D. Bailey, and L. J. van Loon, “Ketone bodies and exercise performance: the next magic bullet or merely hype?” Sports Medicine, vol. 47, no. 3, pp. 383–391, 2017. View at: Publisher Site | Google Scholar
- T. Larsen and N. I. Nielsen, “Fluorometric determination of β-hydroxybutyrate in milk and blood plasma,” Journal of Dairy Science, vol. 88, no. 6, pp. 2004–2009, 2005. View at: Publisher Site | Google Scholar
- N. I. Nielsen, T. Larsen, M. Bjerring, and K. L. Ingvartsen, “Quarter health, milking interval, and sampling time during milking affect the concentration of milk constituents,” Journal of Dairy Science, vol. 88, no. 9, pp. 3186–3200, 2005. View at: Publisher Site | Google Scholar
- B. Stubbs, K. Willerton, S. Hiyama, K. Clarke, and P. Cox, Concomitant Meal Ingestion Alters Levels of Circulating Ketone Bodies following a Ketone Ester Drink, The Physiological Society, London, UK, 2015.
- K. Clarke, K. Tchabanenko, R. Pawlosky et al., “Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects,” Regulatory Toxicology and Pharmacology, vol. 63, no. 3, pp. 401–408, 2012. View at: Publisher Site | Google Scholar
- K. Clarke, K. Tchabanenko, R. Pawlosky et al., “Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate,” Regulatory Toxicology and Pharmacology, vol. 63, no. 2, pp. 196–208, 2012. View at: Publisher Site | Google Scholar
- J. L. Van Hove, S. Grünewald, J. Jaeken et al., “D, L-3-hydroxybutyrate treatment of multiple acyl-CoA dehydrogenase deficiency (MADD),” The Lancet, vol. 361, no. 9367, pp. 1433–1435, 2003. View at: Publisher Site | Google Scholar
- H. Endo, M. Niioka, N. Kobayashi, M. Tanaka, and T. Watanabe, “Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: new insight into the probiotics for the gut-liver axis,” PLoS One, vol. 8, no. 5, Article ID e63388, 2013. View at: Publisher Site | Google Scholar
- K. M. Maslowski and C. R. Mackay, “Diet, gut microbiota and immune responses,” Nature Immunology, vol. 12, no. 1, pp. 5–9, 2011. View at: Publisher Site | Google Scholar
- K. M. Tuohy, L. Conterno, M. Gasperotti, and R. Viola, “Up-regulating the human intestinal microbiome using whole plant foods, polyphenols, and/or fiber,” Journal of Agricultural and Food Chemistry, vol. 60, no. 36, pp. 8776–8782, 2012. View at: Publisher Site | Google Scholar
- J. M. Wong, R. De Souza, C. W. Kendall, A. Emam, and D. J. Jenkins, “Colonic health: fermentation and short chain fatty acids,” Journal of Clinical Gastroenterology, vol. 40, no. 3, pp. 235–243, 2006. View at: Publisher Site | Google Scholar
- M. Velasquez-Manoff, “Gut microbiome: the peacekeepers,” Nature, vol. 518, no. 7540, pp. S3–S11, 2015. View at: Publisher Site | Google Scholar
- O. Kanauchi, T. Iwanaga, K. Mitsuyama et al., “Butyrate from bacterial fermentation of germinated barley foodstuff preserves intestinal barrier function in experimental colitis in the rat model,” Journal of Gastroenterology and Hepatology, vol. 14, no. 9, pp. 880–888, 1999. View at: Publisher Site | Google Scholar
- H. Yadav, J. H. Lee, J. Lloyd, P. Walter, and S. G. Rane, “Beneficial metabolic effects of a probiotic via butyrate-induced GLP-1 hormone secretion,” Journal of Biological Chemistry, vol. 288, no. 35, pp. 25088–25097, 2013. View at: Publisher Site | Google Scholar
- H. J. Kim, P. Leeds, and D. M. Chuang, “The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain,” Journal of Neurochemistry, vol. 110, no. 4, pp. 1226–1240, 2009. View at: Publisher Site | Google Scholar
- Y. Yamawaki, M. Fuchikami, S. Morinobu, M. Segawa, T. Matsumoto, and S. Yamawaki, “Antidepressant-like effect of sodium butyrate (HDAC inhibitor) and its molecular mechanism of action in the rat hippocampus,” World Journal of Biological Psychiatry, vol. 13, no. 6, pp. 458–467, 2012. View at: Publisher Site | Google Scholar
- H. V. Lin, A. Frassetto, E. J. Kowalik Jr. et al., “Butyrate and propionate protect against diet-induced obesity and regulate gut hormones via free fatty acid receptor 3-independent mechanisms,” PLoS One, vol. 7, no. 4, Article ID e35240, 2012. View at: Publisher Site | Google Scholar
- Z. Gao, J. Yin, J. Zhang et al., “Butyrate improves insulin sensitivity and increases energy expenditure in mice,” Diabetes, vol. 58, no. 7, pp. 1509–1517, 2009. View at: Publisher Site | Google Scholar
- K. M. Tuohy, H. M. Probert, C. W. Smejkal, and G. R. Gibson, “Using probiotics and prebiotics to improve gut health,” Drug Discovery Today, vol. 8, no. 15, pp. 692–700, 2003. View at: Publisher Site | Google Scholar
- R. B. Canani, M. Di Costanzo, and L. Leone, “The epigenetic effects of butyrate: potential therapeutic implications for clinical practice,” Clinical Epigenetics, vol. 4, no. 1, p. 4, 2012. View at: Publisher Site | Google Scholar
- A. Alvaro, R. Sola, R. Rosales et al., “Gene expression analysis of a human enterocyte cell line reveals downregulation of cholesterol biosynthesis in response to short-chain fatty acids,” IUBMB Life, vol. 60, no. 11, pp. 757–764, 2008. View at: Publisher Site | Google Scholar
- J. W. Finley, J. B. Burrell, and P. G. Reeves, “Pinto bean consumption changes SCFA profiles in fecal fermentations, bacterial populations of the lower bowel, and lipid profiles in blood of humans,” Journal of Nutrition, vol. 137, no. 11, pp. 2391–2398, 2007. View at: Publisher Site | Google Scholar
- E. E. Canfora, J. W. Jocken, and E. E. Blaak, “Short-chain fatty acids in control of body weight and insulin sensitivity,” Nature Reviews Endocrinology, vol. 11, no. 10, pp. 577–591, 2015. View at: Publisher Site | Google Scholar
- J. Darzi, G. S. Frost, and M. D. Robertson, “Do SCFA have a role in appetite regulation?” Proceedings of the Nutrition Society, vol. 70, no. 1, pp. 119–128, 2011. View at: Publisher Site | Google Scholar
- A. Hague, B. Singh, and C. Paraskeva, “Butyrate acts as a survival factor for colonic epithelial cells: further fuel for the in vivo versus in vitro debate,” Gastroenterology, vol. 112, no. 3, pp. 1036–1040, 1997. View at: Publisher Site | Google Scholar
- J. R. Davie, “Inhibition of histone deacetylase activity by butyrate,” Journal of Nutrition, vol. 133, no. 7, pp. 2485S–2493S, 2003. View at: Publisher Site | Google Scholar
- D. P. Stefanko, R. M. Barrett, A. R. Ly, G. K. Reolon, and M. A. Wood, “Modulation of long-term memory for object recognition via HDAC inhibition,” Proceedings of the National Academy of Sciences, vol. 106, no. 23, pp. 9447–9452, 2009. View at: Publisher Site | Google Scholar
- S. G. Gray, “Epigenetic treatment of neurological disease,” Epigenomics, vol. 3, no. 4, pp. 431–450, 2011. View at: Google Scholar
- J. Segain, D. R. De La Blétiere, A. Bourreille et al., “Butyrate inhibits inflammatory responses through NFkappa B inhibition: implications for Crohn’s disease,” Gut, vol. 47, no. 3, pp. 397–403, 2000. View at: Publisher Site | Google Scholar
- E. L. Vieira, A. J. Leonel, A. P. Sad et al., “Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis,” Journal of Nutritional Biochemistry, vol. 23, no. 5, pp. 430–436, 2012. View at: Publisher Site | Google Scholar
- A. Sabatino, R. Morera, R. Ciccocioppo et al., “Oral butyrate for mildly to moderately active Crohn’s disease,” Alimentary Pharmacology and Therapeutics, vol. 22, no. 9, pp. 789–794, 2005. View at: Publisher Site | Google Scholar
- A. Kotunia, J. Wolinski, D. Laubitz et al., “Effect of sodium butyrate on the small intestine,” Journal of Physiology and Pharmacology, vol. 55, no. 2, pp. 59–68, 2004. View at: Google Scholar
- Y. Furusawa, Y. Obata, S. Fukuda et al., “Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells,” Nature, vol. 504, no. 7480, pp. 446–450, 2013. View at: Publisher Site | Google Scholar
- H. M. Hamer, D. Jonkers, K. Venema, S. Vanhoutvin, F. Troost, and R. J. Brummer, “Review article: the role of butyrate on colonic function,” Alimentary Pharmacology and Therapeutics, vol. 27, no. 2, pp. 104–119, 2008. View at: Publisher Site | Google Scholar
- D. F. MacFabe, N. E. Cain, F. Boon, K. P. Ossenkopp, and D. P. Cain, “Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: relevance to autism spectrum disorder,” Behavioural Brain Research, vol. 217, no. 1, pp. 47–54, 2011. View at: Publisher Site | Google Scholar
- D. F. MacFabe, D. P. Cain, K. Rodriguez-Capote et al., “Neurobiological effects of intraventricular propionic acid in rats: possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders,” Behavioural Brain Research, vol. 176, no. 1, pp. 149–169, 2007. View at: Publisher Site | Google Scholar
- N. Kratsman, D. Getselter, and E. Elliott, “Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model,” Neuropharmacology, vol. 102, pp. 136–145, 2016. View at: Publisher Site | Google Scholar
- M. W. Bourassa, I. Alim, S. J. Bultman, and R. R. Ratan, “Butyrate, neuroepigenetics and the gut microbiome,” Physiological Reviews, vol. 81, pp. 1031–1064, 2001. View at: Google Scholar
- N. I. McNeil, J. Cummings, and W. James, “Short chain fatty acid absorption by the human large intestine,” Gut, vol. 19, no. 9, pp. 819–822, 1978. View at: Publisher Site | Google Scholar
- O. C. Velazquez, H. M. Lederer, and J. L. Rombeau, Butyrate and the Colonocyte. Dietary Fiber in Health and Disease, Springer, Berlin, Germany, 1997.
- G. Sandle, “Salt and water absorption in the human colon: a modern appraisal,” Gut, vol. 43, no. 2, pp. 294–299, 1998. View at: Publisher Site | Google Scholar
- R. Havenaar, “Intestinal health functions of colonic microbial metabolites: a review,” Beneficial Microbes, vol. 2, no. 2, pp. 103–114, 2011. View at: Publisher Site | Google Scholar
- R. B. Canani, G. Terrin, P. Cirillo et al., “Butyrate as an effective treatment of congenital chloride diarrhea,” Gastroenterology, vol. 127, no. 2, pp. 630–634, 2004. View at: Publisher Site | Google Scholar
- J. Butzner, R. Parmar, C. Bell, and V. Dalal, “Butyrate enema therapy stimulates mucosal repair in experimental colitis in the rat,” Gut, vol. 38, no. 4, pp. 568–573, 1996. View at: Publisher Site | Google Scholar
- H. Li, Z. Gao, J. Zhang et al., “Sodium butyrate stimulates expression of fibroblast growth factor 21 in liver by inhibition of histone deacetylase 3,” Diabetes, vol. 61, no. 4, pp. 797–806, 2012. View at: Publisher Site | Google Scholar
- X. Zhang, D. C. Yeung, M. Karpisek et al., “Serum FGF21 levels are increased in obesity and are independently associated with the metabolic syndrome in humans,” Diabetes, vol. 57, no. 5, pp. 1246–1253, 2008. View at: Publisher Site | Google Scholar
- F. Hird and R. Symons, “The mechanism of ketone-body formation from butyrate in rat liver,” Biochemical Journal, vol. 84, no. 1, pp. 212–216, 1962. View at: Publisher Site | Google Scholar
- R. Linskens, X. Huijsdens, P. Savelkoul, C. Vandenbroucke-Grauls, and S. Meuwissen, “The bacterial flora in inflammatory bowel disease: current insights in pathogenesis and the influence of antibiotics and probiotics,” Scandinavian Journal of Gastroenterology, vol. 36, no. 234, pp. 29–40, 2001. View at: Publisher Site | Google Scholar
- R. B. Sartor, “Therapeutic manipulation of the enteric microflora in inflammatory bowel diseases: antibiotics, probiotics, and prebiotics,” Gastroenterology, vol. 126, no. 6, pp. 1620–1633, 2004. View at: Publisher Site | Google Scholar
- A. K. Taggart, J. Kero, X. Gan et al., “(D)-β-hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G,” Journal of Biological Chemistry, vol. 280, no. 29, pp. 26649–26652, 2005. View at: Publisher Site | Google Scholar
- B. Egan and D. P. D’Agostino, “Fueling performance: ketones enter the mix,” Cell Metabolism, vol. 24, no. 3, pp. 373–375, 2016. View at: Publisher Site | Google Scholar
- A. J. Murray and H. E. Montgomery, “How wasting is saving: Weight loss at altitude might result from an evolutionary adaptation,” Bioessays, vol. 36, pp. 721–729, 2014. View at: Google Scholar
- C. C. Blad, K. Ahmed, A. P. Ijzerman, and S. Offermanns, “Biological and pharmacological roles of HCA receptors,” Advances in Pharmacology, vol. 62, pp. 219–250, 2014. View at: Publisher Site | Google Scholar
- S. Offermanns and M. Schwaninger, “Nutritional or pharmacological activation of HCA 2 ameliorates neuroinflammation,” Trends in Molecular Medicine, vol. 21, no. 4, pp. 245–255, 2015. View at: Publisher Site | Google Scholar
- S. Offermanns, “Free fatty acid (FFA) and hydroxy carboxylic acid (HCA) receptors,” Annual Review of Pharmacology and Toxicology, vol. 54, no. 1, pp. 407–434, 2014. View at: Publisher Site | Google Scholar
- S. Offermanns, S. L. Colletti, T. W. Lovenberg, G. Semple, A. Wise, and A. P. Ijzerman, “International union of basic and clinical pharmacology. LXXXII: nomenclature and classification of hydroxy-carboxylic acid receptors (GPR81, GPR109A, and GPR109B),” Pharmacological Reviews, vol. 63, no. 2, pp. 269–290, 2011. View at: Publisher Site | Google Scholar
- J. B. Milder, L. P. Liang, and M. Patel, “Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet,” Neurobiology of Disease, vol. 40, no. 1, pp. 238–244, 2010. View at: Publisher Site | Google Scholar
- M. Storoni and G. T. Plant, “The therapeutic potential of the ketogenic diet in treating progressive multiple sclerosis,” Multiple Sclerosis International, vol. 2015, Article ID 681289, 9 pages, 2015. View at: Publisher Site | Google Scholar
- M. Sandberg, J. Patil, B. D’angelo, S. G. Weber, and C. Mallard, “NRF2-regulation in brain health and disease: implication of cerebral inflammation,” Neuropharmacology, vol. 79, pp. 298–306, 2014. View at: Publisher Site | Google Scholar
- H. C. Huang, T. Nguyen, and C. B. Pickett, “Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription,” Journal of Biological Chemistry, vol. 277, no. 45, pp. 42769–42774, 2002. View at: Publisher Site | Google Scholar
- J. Vriend and R. J. Reiter, “The Keap1-Nrf2-antioxidant response element pathway: a review of its regulation by melatonin and the proteasome,” Molecular and Cellular Endocrinology, vol. 401, pp. 213–220, 2015. View at: Publisher Site | Google Scholar
- N. Wei, D. Yuan, H. B. He et al., “Saponins from Panax japonicas reduces myocardial infarction induced reactive oxygen species production and cardiomyocyte apoptosis via activation of the Nrf-2 pathway,” Advanced Materials Research, vol. 881–883, pp. 339–346, 2014. View at: Publisher Site | Google Scholar
- J. S. Lee and Y. J. Surh, “Nrf2 as a novel molecular target for chemoprevention,” Cancer Letters, vol. 224, no. 2, pp. 171–184, 2005. View at: Publisher Site | Google Scholar
- S. Braun, C. Hanselmann, M. G. Gassmann et al., “Nrf2 transcription factor, a novel target of keratinocyte growth factor action which regulates gene expression and inflammation in the healing skin wound,” Molecular and Cellular Biology, vol. 22, no. 15, pp. 5492–5505, 2002. View at: Publisher Site | Google Scholar
- A. Y. Shih, S. Imbeault, V. Barakauskas et al., “Induction of the Nrf2-driven antioxidant response confers neuroprotection during mitochondrial stress in vivo,” Journal of Biological Chemistry, vol. 280, no. 24, pp. 22925–22936, 2005. View at: Publisher Site | Google Scholar
- J. M. Lee, A. Y. Shih, T. H. Murphy, and J. A. Johnson, “NF-E2-related factor-2 mediates neuroprotection against mitochondrial complex I inhibitors and increased concentrations of intracellular calcium in primary cortical neurons,” Journal of Biological Chemistry, vol. 278, no. 39, pp. 37948–37956, 2003. View at: Publisher Site | Google Scholar
- R. K. Thimmulappa, K. H. Mai, S. Srisuma, T. W. Kensler, M. Yamamoto, and S. Biswal, “Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray,” Cancer Research, vol. 62, no. 18, pp. 5196–5203, 2002. View at: Google Scholar
- R. K. Thimmulappa, C. Scollick, K. Traore et al., “Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide,” Biochemical and Biophysical Research Communications, vol. 351, no. 4, pp. 883–889, 2006. View at: Publisher Site | Google Scholar
- K. U. Tufekci, E. Civi Bayin, S. Genc, and K. Genc, “The Nrf2/ARE pathway: a promising target to counteract mitochondrial dysfunction in Parkinson’s disease,” Parkinson’s Disease, vol. 2011, p. 314082, 2011. View at: Publisher Site | Google Scholar
- X. He, H. Kan, L. Cai, and Q. Ma, “Nrf2 is critical in defense against high glucose-induced oxidative damage in cardiomyocytes,” Journal of Molecular and Cellular Cardiology, vol. 46, no. 1, pp. 47–58, 2009. View at: Publisher Site | Google Scholar
- Y. Tan, T. Ichikawa, J. Li et al., “Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress–induced insulin resistance in cardiac cells in vitro and in vivo,” Diabetes, vol. 60, no. 2, pp. 625–633, 2011. View at: Publisher Site | Google Scholar
- H. Zheng, S. A. Whitman, W. Wu et al., “Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy,” Diabetes, vol. 60, no. 11, pp. 3055–3066, 2011. View at: Publisher Site | Google Scholar
- J. M. Lee, J. Li, D. A. Johnson et al., “Nrf2, a multi-organ protector?” Federation of American Societies for Experimental Biology, vol. 19, no. 9, pp. 1061–1066, 2005. View at: Publisher Site | Google Scholar
- A. Neymotin, N. Y. Calingasan, E. Wille et al., “Neuroprotective effect of Nrf2/ARE activators, CDDO ethylamide and CDDO trifluoroethylamide, in a mouse model of amyotrophic lateral sclerosis,” Free Radical Biology and Medicine, vol. 51, no. 1, pp. 88–96, 2011. View at: Publisher Site | Google Scholar
- S. Yu, T. O. Khor, K. L. Cheung et al., “Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice,” PLoS One, vol. 5, no. 1, Article ID e8579, 2010. View at: Publisher Site | Google Scholar
- H. Nagatomo, Y. Morimoto, A. Ogami et al., “Change of heme oxygenase-1 expression in lung injury induced by chrysotile asbestos in vivo and in vitro,” Inhalation Toxicology, vol. 19, no. 4, pp. 317–323, 2007. View at: Publisher Site | Google Scholar
- S. Suzuki, L. Toledo-Pereyra, F. Rodriguez, and D. Cejalvo, “Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury,” Transplantation, vol. 55, no. 6, pp. 1265–1272, 1993. View at: Publisher Site | Google Scholar
- P. Bowman, Amelioration of Ischemia/Reperfusion Injury During Resuscitation from Hemorrhage by Induction of Heme Oxygenase-1 (HO-1) in a Conscious Mouse Model of Uncontrolled Hemorrhage, DTIC Document, 2012.
- R. Hinkel, B. Petersen, M. Thormann et al., “hHO-1 overexpression in transgenic pigs is cardioprotective after acute myocardial ischemia and reperfsuion,” Circulation, vol. 120, no. 18, p. S1042, 2009. View at: Google Scholar
- T. Yoshida, M. Watanabe, D. T. Engelman et al., “Transgenic mice overexpressing glutathione peroxidase are resistant to myocardial ischemia reperfusion injury,” Journal of Molecular and Cellular Cardiology, vol. 28, no. 8, pp. 1759–1767, 1996. View at: Publisher Site | Google Scholar
- N. S. Dhalla, A. B. Elmoselhi, T. Hata, and N. Makino, “Status of myocardial antioxidants in ischemia–reperfusion injury,” Cardiovascular Research, vol. 47, no. 3, pp. 446–456, 2000. View at: Publisher Site | Google Scholar
- W. Dong, Y. Jia, X. Liu et al., “Sodium butyrate activates NRF2 to ameliorate diabetic nephropathy possibly via inhibition of HDAC,” Journal of Endocrinology, vol. 232, no. 1, pp. 71–83, 2017. View at: Publisher Site | Google Scholar
- X. Chen, W. Su, T. Wan et al., “Sodium butyrate regulates Th17/Treg cell balance to ameliorate uveitis via the Nrf2/HO-1 pathway,” Biochemical Pharmacology, vol. 142, pp. 111–119, 2017. View at: Publisher Site | Google Scholar
- X. Hu, K. Zhang, C. Xu, Z. Chen, and H. Jiang, “Anti-inflammatory effect of sodium butyrate preconditioning during myocardial ischemia/reperfusion,” Experimental and Therapeutic Medicine, vol. 8, no. 1, pp. 229–232, 2014. View at: Publisher Site | Google Scholar
- S. Yano and D. F. Tierney, “Butyrate increases catalase activity and protects rat pulmonary artery smooth muscle cells against hyperoxia,” Biochemical and Biophysical Research Communications, vol. 164, no. 3, pp. 1143–1148, 1989. View at: Publisher Site | Google Scholar
- M. E. Walsh, A. Bhattacharya, K. Sataranatarajan et al., “The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging,” Aging Cell, vol. 14, no. 6, pp. 957–970, 2015. View at: Publisher Site | Google Scholar
Copyright |
| Copyright © 2018 Franco Cavaleri and Emran Bashar. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |

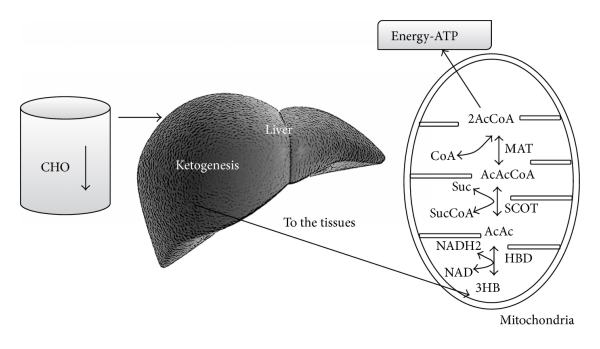 Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.
Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.