Ympäristö- ja mikrobitoksiinien, lääkkeiden, orgaanisten liuottimien ja raskasmetallien vaikutukset multippeliskleroosin puhkeamiseen ja etenemiseen
Kliinisten tieteiden laitos, College of Medicine, immuno-onkologinen ryhmä, Sharjahin lääketieteellisen tutkimuksen instituutti (SIMR), Sharjahin yliopisto, Sharjah, United Arab Emirates
28. tammikuuta 2019 / Tarkistettu: 13. helmikuuta 2019 / Hyväksytty: 28. helmikuuta 2019 / Julkaistu: 5. maaliskuuta 2019
Tiivistelmä
Multippeliskleroosi on nuorten aikuisten yleisin neurologinen sairaus. Se aiheuttaa sairastuneen ja tämän läheisten elämään sosiaalisia, terveydellisiä ja taloudellisia haasteita. MS-tauti lisää sosiaali- ja terveyspalvelujen taloudellista taakkaa. Mikä multippeliskleroosin aiheuttaa?
MS-taudin syistä on esitetty monenlaisia väittämiä. Osa väitteistä on hyvin perusteltuja. Sen sijaan eräät MS-taudin syistä esitetyt hypoteesit sotkevat korrelaation ja kausaation keskenään.
Multippeliskleroosi on osoittautunut lääketieteellisesti vaikeaksi ongelmaksi. Kyseessä on oireiltaan ja patologialtaan monimutkainen ja monitekijäinen sairaus. Tautiin vaikuttavia geenimuunnoksia tunnetaan toista sataa. Näiden geenimuutosten erilaiset variantit vaikuttavat eri tavoin eri potilaisiin. Osalla sairastuneista tauti etenee aggressiivisesti ja nopeasti, kun taas toiset voivat elää lähes oireettomasti vuosikymmeniä.
Kaikille MS-tautia sairastaville potilaille sopivaa farmakologista hoitoa on vaikea kehittää, koska tautiin liittyy valtavasti geneettisiä, epigeneettisiä ja ekologisia muuttujia. Multippeliskleroosiin sairastuminen on surullisten sattumusten sarja.
Yritykset ymmärtää MS-taudin etiopatologiaa eivät ole löytäneet vedenpitävää vastausta peruskysymykseen: Mikä MS-taudin aiheuttaa?
Yleisestä epätietoisuudesta syntyy helposti ajatus, että taudin aiheuttaja tunnetaan, mutta sitä ei kerrota, koska MS-potilaat ovat hyvä tulojen lähde lääketeollisuudelle. Se ei varmaankaan ole totta.
Kymmeniä tai satoja taudin etiologiaan liittyviä geenimuunnoksia, epigeneettisiä vaikuttajia ja ympäristötekijöitä on tunnistettu, mutta selkeää vastausta sairauden syistä ja siihen vaikuttavista solutason mekanismeista ei täysin ymmärretä. Tehostuneet tutkimusmenetelmät kuitenkin näkevät tarkemmin ja syvemmälle ihmisen keskushermostoon. Molekyylibiologian ja solujen aineenvaihdunnan tutkimuksen sekä tehostuneiden tutkimusvälineiden myötä käsityksemme taudin syistä ja mekanismeista tarkentuu.
Tässä katsauksessa tutustutaan MS-autoimmuniteetin taustalla vaikuttaviin geneettisiin, epigeneettisiin ja ekologisiin tekijöihin. Katsauksen pääpaino kohdistetaan sellaisten toksiinien, kemikaalien tai lääkkeiden käyttöön, jotka voivat laukaista, muuttaa, hidastaa tai ehkäistä MS-tautia ja siihen liittyviä oireita.
1. Johdanto
Multippeliskleroosi (MS) vaurioittaa keskushermostoa (CNS). Tauti johtaa aivojen ja selkäytimen valkeassa ja harmaassa aineessa primaariseen multifokaaliseen demyelinaatioon ja diffuusiin neurodegeneraatioon [1].
Multifokaalinen demyelinaatio viittaa monesta paikasta alkaviin viejähaarakkeita suojaavien myeliinituppien vaurioihin aivojen valkeassa aineessa (demyelinaatio). Diffuusilla neurodegeneraatiolla tarkoitetaan epätarkkarajaisia ja epäselviä soluvauriota ja -surkastumia erityisesti aivojen harmaassa aineessa (neurodegeneraatio).
Suurin osa MS-tautiin sairastuneista sairastaa aaltomaisesti etenevää, eli relapsoivaa-remittoivaa tautimuotoa, jossa taudin pahenemisvaiheita seuraa toipumis- ja vakausjaksot [2].
Sekä patologiset että radiologiset havainnot viittaavat neuroinflammation ja neurodegeneraation varhaiseen rinnakkaisvaikutukseen taudin patogeneesissa [3].
MS-tauti on eräs yleisimmistä nuorten aikuisten invalidisoitumista aiheuttavista neurologisista häiriöistä [4]. Globaalisti multippeliskleroosia sairastaa arviolta 2,5 miljoonaa ihmistä. Tautiin sairastuvat yleensä nuoret aikuiset. Naisilla tautia esiintyy noin kaksi kertaa enemmän kuin miehillä [5].
Multippeliskleroosi aiheuttaa merkittävän henkilökohtaisen ja sosioekonomisen taakan, sillä suurin osa potilaista tarvitsee apuvälineitä, farmakologista hoitoa, seurantaa ja henkilökohtaista apua [6].
Multippeliskleroosi on arvaamaton sairaus, jonka potilaskohtaisesta etenemisestä voidaan antaa vain tilastollisia suuntaviivoja. Yleisesti ottaen oireiden kirjo ja taudin eteneminen ovat yksilöllisiä. Multippeliskleroosiin vaikuttavat geneettiset, epigeneettiset ja ekologiset tekijät voivat kiihdyttää tai hidastaa taudin kulkua [7].
Vaikka kokeellinen malli taudista on olemassa, se ei selitä taudin vaihtelevia kliinisiä, patologisia tai immunologisia piirteitä [8].
MS-taudin hoito- ja terapiamenetelmät ovat muuttuneet vuosien varrella. Taudin oireita ja pahenemisvaiheita pyritään hillitsemään immuunijärjestelmän toimintaa modifioivilla ja oireenmukaisilla lääkehoidoilla. Toimintakykyä ylläpidetään erilaisilla kuntoutushoidoilla, joiden toivotaan hidastavan sairastuneen invalidisoitumista [9].
Multippeliskleroosin syistä tiedetään edelleen aivan liian vähän. Ymmärrys taudin etiologiasta on pysynyt paljolti samana kuin William Boydin aikana. Boyd totesi vuonna 1958:
”Aikaa ja rahaa, joka on käytetty multippeliskleroosin syy-tekijöiden määrittämiseen, ei voida laskea … tulos on ollut nolla” [10].
Tietämättömyys taudin syistä on omiaan lisäämään villejä arvauksia. Joissain tapauksissa villit arvaukset perustuvat tieteellisesti validiin ja loogisesti koherenttiin päättelyyn. Tällaisissa tapauksissa voidaan havaita vahva korrelaatio jonkin patogeenin tai ymäristötekijän ja MS-taudin väliltä, vaikka kausaatiota ei pystyttäisi osoittamaan.
Esimerkiksi D-vitamiinin puute ja eurooppalainen perimä korreloivat vahvasti MS-taudin kanssa. On saatu vahvaa näyttöä siitä, että odottavan äidin matalat D-vitamiinitasot altistavat lapsen myöhemmin kehittyvälle MS-taudille. Ascherio yms. on tutkinut tätä korrelaatiota varsin kattavasti. Toisaalta on myös havaittu, että odottavan äidin Epstein-Barr-infektio (mononukleoosi) ja sikiön kohonneet IgG-vasta-ainepitoisuudet korreloivat lapsen kasvaneen MS-riskin kanssa.
”Interpretation: Offspring of mothers with high viral capsid antigen IgG during pregnancy appear to have an increased risk of MS. The increase in MS risk among women with elevated prediagnostic EBNA-1 IgG levels is consistent with previous results.”
2. Kateenkorva, T-solut & immunologinen toleranssi
Immuunivälitteisellä patogeneesillä on keskeinen rooli MS-taudissa [11]. Huolimatta siitä, että veri-aivoesteen tulisi estää immuunisolujen pääsy keskushermostoon, multippeliskleroosia sairastavien keskushermostosta löydetään merkkejä T-soluista ja B-solujen tuottamia immunoglobuliineja. MS-taudissa veri-aivoeste vuotaa ja päästää lävitseen immuunisoluja [12]. Mekanismi tunnetaan, mutta syistä on vain valistuneita arvauksia.
T-solut kypsyvät ja erilaistuvat kateenkorvasssa (thymus). Kypsyminen tuottaa valtavasti eri antigeeneille herkistyneitä T-soluja. Samalla kehittyy aina jonkin verran autoreaktiivisia T-soluja. Immunologisen toleranssin negatiivisen selektion pitäisi ohjata nämä omille kudoksille herkistyneet T-solut apoptoosiin, eli ohjattuun solukuolemaan.
Autoimmuunitaudeissa myös immunologinen toleranssi falskaa. Immunologisen toleranssin negatiivinen selektio ei jostain syystä poista autoreaktiivisia T-soluja, minkä seurauksena verenkiertoon vapautuu elimistön omille soluille herkistyneitä immuunisoluja. Nämä voivat kohdistaa aktivaationsa kehon omia kudoksia, kuten myeliiniä vastaan.
Kateenkorva ja immunologinen toleranssi vaikuttavat autoimmuunitautien patogeneesiin.
Immunologinen toleranssi tarkoittaa immuunijärjestelmän kykyä olla reagoimatta elimistön omiin kudoksiin ja harmittomiin vieraisiin rakenteisiin. Toleranssin häiriöt voivat johtaa allergioihin tai autoimmuunitauteihin.
Immunologinen toleranssi jaetaan sentraaliseen ja perifeeriseen toleranssiin, ja se on hankinnaisen eli adaptiivisen immuunijärjestelmän ominaisuus. Lymfosyytteihin kuuluvien CD4-positiivisten T-solujen merkitys on tärkeä, sillä niiden erilaistuminen Th1-, Th2- tai Th17-efektorisoluiksi tai säätelijä-T-soluiksi ohjaa koko immuunijärjestelmän toimintaa.
Th1-tyypin immuunivaste on soluvälitteinen. Th2-vasteessa korostuu B-lymfosyyttien IgE-luokan vasta-aineiden tuotanto. Jälkimmäisen yhteys allergioihin tunnetaan hyvin. Th17-vaste tukee neutrofiilien toimintaa ja vahvistaa epiteelejä. Säätelijä-T-solut puolestaan pitävät yllä toleranssia estämällä haitallisia immuunivasteita ja huolehtivat immuunivasteen sammuttamisesta, kun taudinaiheuttaja on saatu hävitetyksi elimistöstä.
T-solujen toiminta perustuu T-solupopulaation kykyyn tunnistaa peptidejä mistä tahansa vieraasta tunkeutujasta mutta olla samalla reagoimatta elimistön omiin rakenteisiin. Yksittäinen T-solu tunnistaa reseptorillaan vain tietyn peptidin sitoutuneena tiettyyn MHC-molekyyliin, mutta T-solupopulaation suuri koko ja reseptorien monimuotoisuus takaavat sen, että minkä tahansa vieraan proteiiniin pilkkomistuotteisiin reagoi ainakin muutama T-solu. Nämä piirteet ovat peräisin T-solujen maturaatiosta kateenkorvassa.
Kateenkorvassa lymfosyyttien esiasteiden kehitys T-soluiksi alkaa T-solureseptoria koodaavien geenisegmenttien uudelleenjärjestelyllä eli rekombinaatiolla. Koska geenisegmenttejä on suuri määrä ja yhdistelyyn liittyy sattumanvaraista epätarkkuutta, rekombinaation tuloksena syntyy antigeenispesifisyydeltään erittäin monimuotoinen kehittyvien T-solujen populaatio. Onnistuneen beetaketjun rekombinaation jälkeen solut jakautuvat muutaman kerran ja alkavat sitten ilmentää pinnallaan CD4- ja CD8-proteiineja.
Positiivinen selektio on välttämätön T-solureseptorin toimivuuden testaamiseksi, mutta sentraalinen toleranssi perustuu reseptorin autoreaktiivisuuden testaamiselle prosessissa, jota kutsutaan negatiiviseksi selektioksi.
Kateenkorvan epiteelisolut tuottavat AIRE-geenin ohjaamina myös sellaisia proteiineja, jotka normaalisti esiintyvät vain tietyissä elimistön osissa, kuten hermostossa tai umpirauhasissa. Näistä elimistön omista proteiineista pilkottuja peptidejä esitellään MHC-molekyyleihin sitoutuneina kehittyville T-soluille. Jos kehittyvä T-solu sitoutuu voimakkaasti oman MHC-molekyylin ja oman peptidin yhdistelmään, solu tulee negatiivisesti valikoituneeksi ja kuolee apoptoottisesti.
Negatiivinen selektio karsii T-solupopulaatiosta voimakkaasti autoreaktiiviset solut ja synnyttää sentraalisen toleranssin. – Duodecim
Tieteellinen yhteisö ei kuitenkaan ole yksimielinen siitä, onko MS klassisen määritelmän mukaan ensisijaisesti autoimmuunisairaus, tulehdustekijöihin liittyvä demyelinoiva tauti, johon liittyy autoimmmuuni-ilmentymiä, mitokondrioiden häiriintyneeseen toimintaan liittyvä neurodegeneratiivinen sairaus vai kaikkia näitä tai jotain näiden väliltä [3].
Havainto, että puolet MS-taudin immuunitoimintaan liittyvistä geneettisistä muunnoksista liittyy myös muiden autoimmuunisairauksien patologiaan, tukee autoimmuunimallia [6,7].
Viime aikoina tautia on kutsuttu prototyyppiseksi autoimmuunikeskushermostosairaudeksi [8, 9, 10], jossa autoimmuunivälitteiset myeliinivauriot liittyvät esimerkiksi epigeneettiseen alttiuteen [11]. Tällaisessa autoimmuunimallissa autoreaktiiviset ja adaptiiviset immuunisolut tunkeutuvat keskushermostoon ja voimistavat keskushermoston aksonivaurioita [12].
CD4+ -T-lymfosyyttejä pidetään laajalti tärkeimpinä toimijoina MS-taudin patogeneesissä [7]. Funktionaalisesti muuttuneiden T-auttajasolujen (alatyypit Th1 ja Th17) ja Treg-solujen sekä muiden leukosyyttipopulaatioiden, kuten luonnollisten tappajasolujen (NK) löydökset MS-tautia sairastavien selkäydinnesteestä (CSF) on hyvin dokumentoitu [13].
Näiden lymfosyyttien toiminnassa havaitaan toiminnallisia vikoja T- ja B-säätelyalaryhmissä sekä tulehdusta edistävää (proinflammatorista) profiilia [14]. Autoreaktiiviset Th17-solut voivat läpäistä veri-aivoesteen (BBB) solujen tiukkoja liitosproteiineja ja endoteelisoluja heikentävien IL-17- ja IL-22-sytokiinien avustamana. Tämä johtaa neutrofiilien aktivaatioon ja hermosolujen vaurioihin [15].
Patogeenisillä Th17-soluilla on heikko FasL-ilmentyminen, joten ne voivat välttää ohjelmoidun solukuoleman (apoptoosin) ja säilyä tulehtuneissa kohdissa [16]. On arveltu, että virusinfektion yhteydessä tapahtuva immuunisolujen aktivaatio voi tuottaa sellaisia autoreaktiivisia ja mahdollisesti enkefalitogeenisiä* T-auttaja (Th) -1/17 -soluja, joita kehittyy selkäydinnesteeeseen (CSF) MS-pahenemisvaiheen jälkeen [14].
*Enkefalitogeenisellä tarkoitetaan myeliiniin aktivoituvaa.
Näiden tunnistettujen vaikuttajien lisäksi on löydetty uusia immuunisoluja, kuten Interleukiini (IL) -9:n tuottamat CD4+ T-auttajasolut ’Th9’ [17] ja T-auttaja 22 ’Th22’ -solut [18], jotka helpottavat taudin aktivoitumista ja etenemistä. Hiljattain havaittiin myös, että MS-vaurioissa CD8+ T-solut näyttelevät suurempaa roolia kuin CD4+ T-solut. Immuunisolujen klooneja havaittiin sairastuneiden veressä ja selkäydinnesteessä vielä useiden vuosien jälkeen [7].
B-solupopulaatioiden tutkimus MS-plakkeissa paljasti kloonisesti laajentuneiden B-lymfosyyttien kertymisen, mikä osoittaa B-solujen, vasta-aineiden ja sen komplementin keskeisen roolin demyelinaatioprosessissa [19].
Lisäksi dendriittisolut, jotka toimivat antigeeniä esittelevinä soluina (APC) sen lisäksi, että ne ovat efektorisoluja neuro-tulehduksessa, pahentavat MS-patologiaa, mutta APC:n rooli MS-taudin patogeneesissä tunnetaan epätäydellisesti [7].
Näiden havaintojen perusteella ympäristötekijöiden vaikutus geneettisen alttiuden omaaviin ihmisiin on tärkeää, koska ympäristötekijät voivat laukaista MS-tautiin johtavan vahingollisen kaskadin (reaktioketjun).
Vaikka MS ei ole perinnöllinen sairaus, MS-taudin perhetapausten ryhmittyminen on yleistä sellaisten ensimmäisen asteen sukulaisten keskuudessa, joilla on yhtäläisyyksiä tärkeimmässä histokompatibiilikompleksissaan (MHC), kuten HLA DR15 / DQ6-alleeli, interleukiini-2-reseptori-alfa-geenin alleelit ’IL2RA’ ja interleukiini-7-reseptorialfa-geeni ’IL7Ra’ [20]. Lisäksi jotkut MS-potilaat osoittivat spesifisiä yhden nukleotidin polymorfismeja tällaisissa geeneissä [21].
On havaittu, että nämä geneettiset polymorfismit liittyvät immuunijärjestelmään ja voivat siten lisätä autoimmuunisairauksien alttiutta. Geneettinen taipumus selittää kuitenkin vain murto-osan taudin riskeistä [22]. Vaikka tällä hetkellä yli sadan geenin tiedetään lisäävän MS:n riskiä, ne vaikuttavat vain marginaalisesti [23]. Siksi MS-taudin kehittymiselle on oltava kattavampi selitys.
Alkuperäisvaikutus (eli alleelin fenotyyppinen vaikutus riippuu siitä, onko se peritty yksilön äidiltä vai isältä). MS-taudin korkeampi esiintyvyys naisilla liittyy epigeneettiseen X-kromosomien inaktivaatioon [21], mikä osoittaa epigeneettisten muutosten tutkimisen tärkeyden tällaisilla potilailla. MS-taudissa epigeneettisten mekanismien on osoitettu vaikuttavan T-solutoimintoihin, joissa histoniasetyloinnin raportoitiin esiintyvän keskushermoston valkeassa aineessa, hypermetylaation oligodendrosyyttien eloonjäämisgeeneissä ja hypo-metylaation proteolyyttisissä prosessointigeeneissä [24].
Lisäksi epidemiologiset tutkimukset ovat osoittaneet geneettisen alttiuden ja ympäristön välisen vuorovaikutuksen moduloivan immuunijärjestelmän epigenomia [21]. Tällaiset epigeneettiset mekanismit reagoivat helposti ympäristötekijöihin [25,26]. Vaikka geneettiset tai epigeneettiset tekijät voivat johtaa autoimmuniteettiin, mekanismi tunnetaan vielä heikosti.
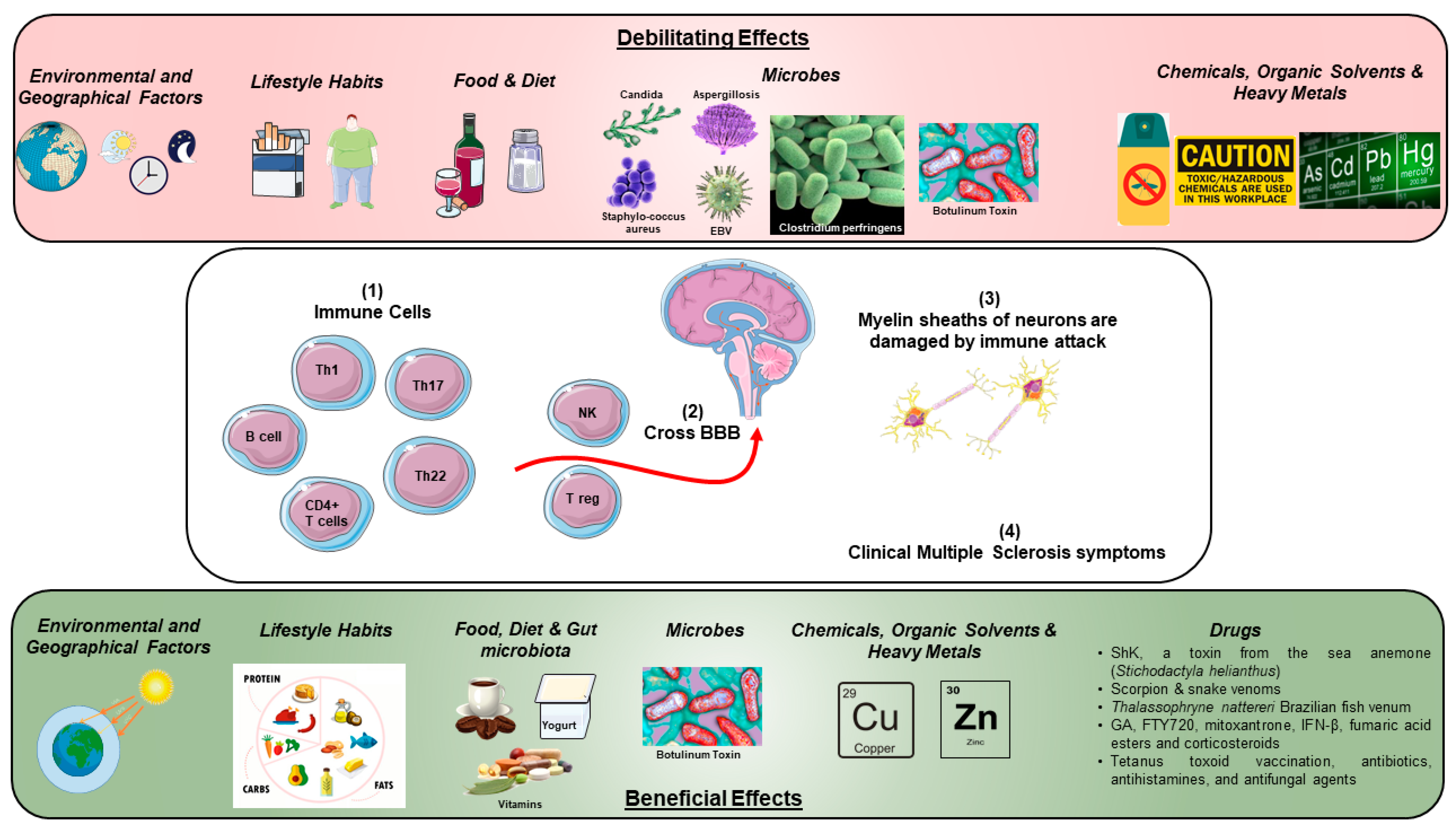
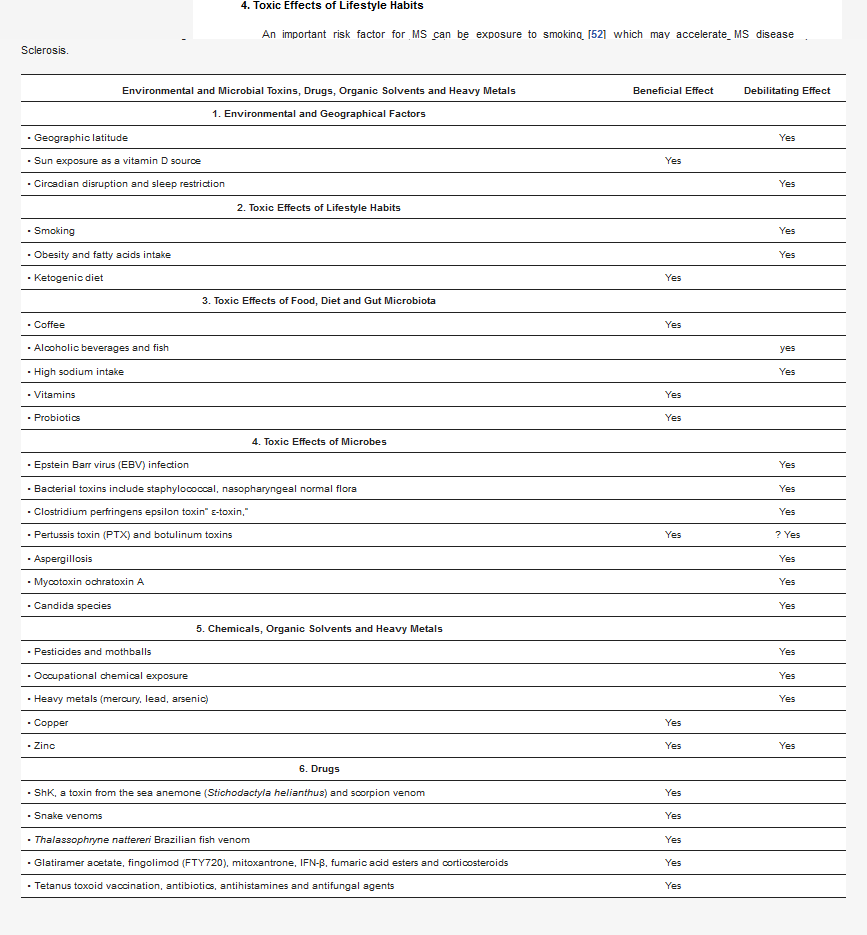
Yleinen oletus on, että ihmisellä on muuttumaton genomi ja useita muuttuvia epigenomeja. Yhteenveto sellaisista taudin laukaisijoista, joilla voi olla hyödyllisiä ja heikentäviä vaikutuksia multippeliskleroosin puhkeamiseen ja etenemiseen, on lueteltu taulukossa 1 ja kuvattu kuvassa 1.
Kuva 1. Ympäristö- ja mikrobitoksiinien, lääkkeiden, orgaanisten liuottimien ja raskasmetallien hyödylliset ja heikentävät vaikutukset multippeliskleroosin puhkeamiseen ja etenemiseen.
Taulukko 1. Ympäristö- ja mikrobitoksiinien, lääkkeiden, orgaanisten liuottimien ja raskasmetallien hyödylliset ja heikentävät vaikutukset multippeliskleroosin puhkeamiseen ja etenemiseen.
3. Ympäristön ja maantieteellisten tekijöiden vaikutukset
Ympäristövaikutukset muuttavat taudin riskiä ja etenemistä mahdollisesti epigeneettisten vaikutusten välityksellä säätelemällä immuunivastetta ylös- tai alaspäin ja vaikuttamalla hermoston kehitykseen [23, 27].
Altistuminen orgaanisille liuottimille ja alkoholille, runsas kahvin kulutus [22,28 ], infektiot, auringonvalo / D-vitamiini ja tupakointi vaikuttavat MS-tautiin ja sen etenemiseen [29], mutta näiden syy-yhteyden osoittamiseksi ei ole vielä riittävästi tutkimuksia [30].
MS on jakautunut epätasaisesti. Esiintyvyys kasvaa asteittain maantieteellisen leveyspiirin mukaan [31]. Monet ovat epäilleet, että ympäristön saasteiden ja MS-taudin esiintyvyyden välillä olisi positiivinen yhteys. Ympäristöelementtien roolia taudin kehittymisessä on tutkittu laajasti, mutta minkäälaiseen loppupäätelmään ympäristötoksiinien roolista ei olla päästy [32].
MS-taudin maantieteellisestö epätasaisesta jakautumisesta osoittaa mm. se, että Key Westissä Floridassa on epätavallisen suuri multippeliskleroosin esiintyvyys [33]. MS on myös selvästi yleisempää Ison-Britannian ja Pohjois-Irlannin pohjoisosissa ja Skotlannin saaristossa kuin Englannissa ja Walesissa [34]. Tämä viittaa vahvaan yhteyteen maantieteen ja multippeliskleroosin esiintyvyyden välillä [35]. Yhteyttä tukee edelleen Kanadassa tehty tutkimus, jossa osavaltioiden MS-esiintyvyys vaihtelee alueittain, mikä viittaa siihen, että erot taudin esiintyvyydessä voivat johtua paikallisista ympäristötekijöistä [36].
Toisaalta tutkimukset ovat osoittaneet, että MS:n esiintyvyyden pohjoinen / etelä-vaihtelu voi johtua muutoksesta populaatioiden geneettisessä alttiudessa MS-tautiin [37]. Monien ympäristötekijöiden joukossa auringonvalo D-vitamiinin lähteenä näyttelee keskeistä roolia MS-taudin patogeneesissä.
Epidemiologisissa väestötutkimuksissa on johdonmukaisesti osoitettu, että MS-taudin riski on suurempi alueilla, joilla auringonvalo on vähäistä. Ascherio et al. ovat osoittaneet, että odottavan äidin ja sikiön matalat D-vitamiinitasot kasvattavat syntyvän lapsen riskiä sairastua myöhemmin MS-tautiin [38,39]. Johdonmukaiset ja yhdenmukaiset havainnot viittaavat siihen, että D-vitamiinin puutos on MS-taudin riskitekijä [40 ]. Tämä tukee ajatusta D-vitamiinin saannin suojaavista vaikutuksista MS-taudin riskiä laskevana tekijänä [41].
Etelä-Amerikassa autoimmuunitauteja hoidetaan Coimbra-protokollalla, joka perustuu hyvin korkeisiin D-vitamiinin annostuksiin. Sovelsin itseeni Coimbra-protokollaa noin vuoden ajan, mutta en osaa sanoa oliko siitä mitään hyötyä tai haittaa, koska mitään vertailukohtaa ei ole. Viimeisten 5-7 vuoden aikana oma PPMS-tautini on kuitenkin edennyt hyvin hitaasti.
Tutkimusten mukaan D3-vitamiinihoito parantaa kliinisiä oireita kokeellisessa autoimmuunisen enkefalomyeliitin “EAE” -hiirimallissa [42]. On osoitettu, että vastasyntyneen D-vitamiinin matalat pitoisuudet liittyvät lisääntyneeseen MS-riskiin [43]. Esimerkiksi marraskuussa syntyneillä MS-taudin esiintyvyys on vähentynyt merkittävästi, mikä liittyy vastasyntyneiden korkeaan D-vitamiinialtistukseen raskauden kolmannen kolmanneksen aikana suojaavana tekijänä multippeliskleroosia vastaan [44]. D-vitamiinireseptorin (VDR) ilmentyminen on lisäksi estetty MS-taudissa. Ympäristön, genetiikan ja epigeneettisten tekijöiden tiedetään vaikuttavan D-vitamiinin aineenvaihduntaan [45].
D-vitamiinia sitovan proteiinin lisääntyminen MS-potilaiden seerumissa pahentaa taudin patofysiologiaa [46]. On havaittu, että ultraviolettisäteily voi heikentää Th1-välitteisiä immuunivasteita [31] tai vähentää immunostimulatorisen neurohormoni melatoniinin eritystä käpylisäkkeestä [47].
Vuorokausirytmin häiriöt ja vähäinen uni voivat häiritä melatoniinin eritystä ja siten lisätä tulehdusta edistäviä reaktioita. Tämä saattaa antaa selityksen tutkimuksille, jotka yhdistävät MS-taudin, iän ja vuorokausirytmin [48,49]. Tutkimukset osoittavat tilastollisesti merkitsevän yhteyden nuorena tehdyn vuorotyön ja MS-riskin välillä [50,51]. Elämäntapa- ja ympäristötekijät ovat keskeisiä MS-taudin riskiin vaikuttavia tekijöitä [22].
Tästä syystä lisätutkimuksissa olisi keskityttävä MS-taudin mahdollisten juurien selvittämiseen tutkimalla potilaiden elämäntavat (ruokavalio, liikunta jne.) ja niiden vaikutus patogeenisiin tapahtumiin [29].
4. Elintapatottumusten vaikutukset
Tupakointi ja tupakansavu on merkittävä MS-taudin riskiä lisäävä tekijä [52]. Havaintojen mukaan tupakointi nopeuttaa MS-taudin etenemistä ja invalidisoitumista [53]. Tupakoinnin uskotaan kiihdyttävän RRMS-tautimuodon etenemistä toissijaisesti progressiiviseksi MS-taudiksi (SPMS) [54].
Tupakointiin assosioituva riski lisääntyy edelleen HLA-DRB1 * 15-geenimuunnoksen omaavilla tupakoitsijoilla johtuen spesifisestä T-soluvasteesta savulle, joka voi pahentaa geneettisesti säänneltyä makrofagivastetta [55]. Tupakointi ja tupakan savulle altistuminen on osoitettu MS-taudin riskitekijäksi [56].
Jos MS-tautia esiintyy suvussa, tupakointi lisää selvästi sairastumisen riskiä. Sukulaisen sairastuminen MS-tautiin on varoitusmerkki, jonka jälkeen tupakoinnin mielekkyyttä on syytä arvioida uudelleen [57]. Toisaalta on olemassa vahvaa näyttöä puberteetti-ikäisten lihavuuden roolista riskitekijänä, joka lisää MS-tautiin sairastumisen riskiä [22,56].
Edelliset havainnot selittävät omaa sairastumistani. Olin murrosiässä ylipainoinen, aloitin tupakoinnin varhain ja suvussa esiintyy MS-tautia.
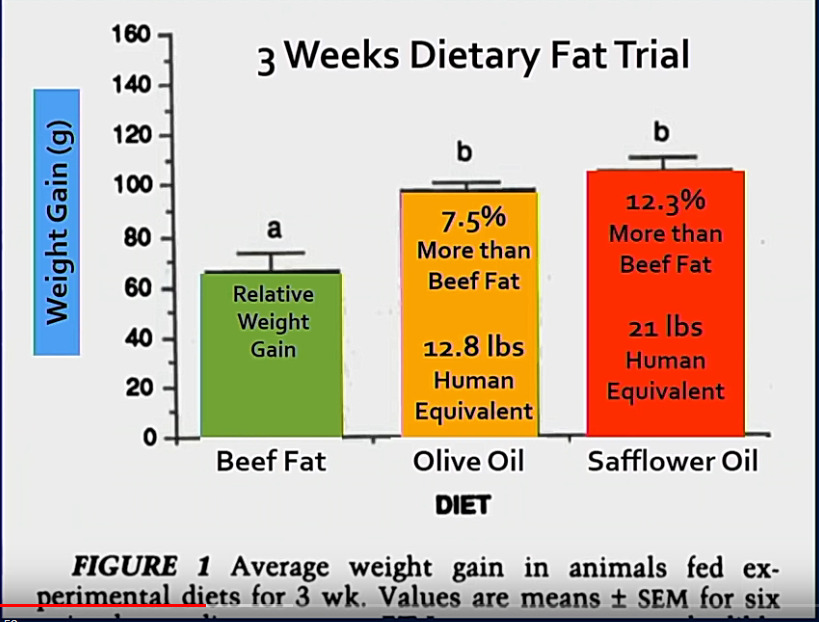
MS-taudin esiintyvyyden. vakavuuden ja rasvahappojen saannin välillä on myös dokumentoituja yhteyksiä [58]. Monityydyttymättömät rasvahapot (PUFA) vähensivät relapsien esiintyvyyttä kahden vuoden seurannan aikana [59].
Lisäksi ketogeenisellä ruokavaliolla voi olla suojaavia vaikutuksia, mikä todennäköisesti johtuu solutason vaikutuksista immuunivasteeseen ja oksidatiivista stressiä hillitsevästä vaikutuksesta [60]. Ketogeenisen ruokavalion positiivisista vaikutuksista MS-taudin terapiana olen käsitellyt näissä tutkimuskatsauksissa:
Pilottitutkimus: Ketogeeninen ruokavalio ja RRMS
Ketogeeninen ruokavalio ja PPMS
5. Ravintoaineiden ja suoliston mikrobiomin vaikutukset
MS-tutkimuksen eläinmalleissa havaittiin, että runsas kahvin kulutus saattaa vähentää MS-taudin riskiä tukahduttamalla tulehdusta edistävien sytokiinien tuotantoa [61] ja kofeiinin neuroprotektiivisten ominaisuuksien vuoksi [62]. Kahvi on myös monien suomalaisten tärkein polyfenoleiden lähde. Kahvin on havaittu tehostavan kognitiivisia kykyjä ja suojaavan maksaa.
Alkoholi ja kala liittyvät invalidisoitumisen etenemiseen relapsoivassa remittoivassa MS-taudissa [63]. Myös suuri natriumin saanti voi pahentaa sairauden aktiivisuutta sekä kliinisiä ja radiologisesti havaittavia oireita [64].
Kalan negatiivinen vaikutus MS-taudin oireisiin on herättänyt keskustelua, koska kala mielletään terveelliseksi. Tutustumatta viitattuun tutkimukseen uskon kalan korkeiden raskasmetalli- yms. toksiinipitoisuuksien voivan selittää proinflammatorisia vaikutuksia, jotka pahentavat MS-taudin oireita. Kalan omega-3-rasvahapoilla on tiettävästi neutraali tai oireita hillitsevä vaikutus.
D3-vitamiinin puutteellinen saanti on MS-taudin riskitekijä. Muiden vitamiinien tai mineraalien vaikutuksista MS-taudin puhkeamiseen ei ole riittävää näyttöä [65]. Ruokavalio, D3-vitamiinin puutos, tupakointi ja alkoholin runsas käyttö vaikuttavat suoliston mikrobiomin koostumukseen [66]. Suolen mikrobiomi määritellään kaikeksi mikrobipitoisuudeksi, mukaan lukien geenit, proteiinit ja aineenvaihduntatuotteet suolistossa tiettynä ajankohtana [67]. Mahdolliset häiriöt suolen mikrobiomissa tai ns. ”dysbioosi” assosioituvat moniin sairauksiin [68].
MS-potilailla on havaittu vaurioita suoliston mikrobiomin koostumuksessa. Suoliston mikrobiomin terveys ja lajikirjo voivat näytellä merkittävää roolia MS-taudin patogeneesissä [69]. Lisäksi suoliston dysbioosin on havaittu lisäävän suoliston ja veri-aivoesteen läpäisevyyttä mikrobiomi-suolisto-aivo-akselin välityksellä. Siihen voidaan vaikuttaa probioottien saannilla [70]. Tässä suhteessa parantunut hygienia vaikuttaa autoimmuunisairauksiin, mikä korostaa suolistoflooran roolia ja vaikutusta EAE:n kehittymiseen MS-taudin hiirimallissa [71].
6. Mikrobien vaikutukset
Veden vaurioittamissa ympäristöissä elää homesieniä sekä gram-negatiivisia ja gram-positiivisia bakteereja [72]. Tällainen ympäristö sisältää monia biotoksiineja, jotka voivat johtaa MS-tyyppisten sairauksien klusteriin [73]. Myös monilla tartunta-aineilla on merkitys MS-taudin puhkeamisessa [74], koska erilaiset virukset voivat laukaista MS-taudin ja sitä muistuttavia tulehduksellisia demyelinoivia sairauksia [75].
Esimerkiksi primaarinen Epstein Barr-infektio voi laukaista MS-taudin kehittymisen geneettisen alttiuden omaavilla nuorilla aikuisilla [76]. Bakteeritoksiinit, stafylokokki-bakteerit, nenänielun normaalin mikrobiston häiriöt ja monet muut tekijät voivat vaurioittaa immuniteettia ja aiheuttaa vaurioita hermostossa.
Stafylokokkitoksiinit stimuloivat ihmisen T-lymfosyyttejä, mikä johtaa myeliinin autoantigeenien, myeliinin emäksisen proteiinin ja proteolipidipeptidin aktivoitumiseen. Tämä aktivoi reaktiivisia T-lymfosyyttejä, jotka myötävaikuttavat demyelinoivaan prosessiin [77]. Tämä on mahdollista, koska selkäydinneste (CSF) ja solunulkoinen nestekierto ovat yhteydessä kaksisuuntaisesen reitin kautta. Nenänielun infektiotuotteet voivat valua keskushermostoon ja vaikuttaa aivokalvojen immuunisoluihin, mikä puolestaan voi johtaa keskushermostoon syntyviin vaurioihin [78].
Toinen MS-tautiin mahdollisesti vaikuttava bakteeritoksiini on Clostridium perfringens epsilon-toksiini, eli ”e-toksiini”. E-toksiini sitoutuu valkean aineen myeliiniin, jolloin myeliini turpoaa ja vaurioituu. Blanch M et al. tunnisti myeliini- ja lymfosyyttiproteiinin (MAL) avainproteiiniksi, joka välittää e-toksiinin sytotoksisen vaikutuksen tulehduksellisissa autoimmuunisairauksissa, kuten MS [79]. Lisäksi e-toksiini voi läpäistä veri-aivoesteen ja sitoutua myeliiniin [80,81]. Tämä johtaa oligodendrosyyttien ja/tai myeliinin vaurioitumiseen [82].
Wagley et al. osoitti Clostridium perfringens ε-toksiinin korreloivan multippeliskleroosin sairastamisen kanssa Yhdysvaltain populaatiossa [83]. Pertussis-toksiini (PTX) ja botuliinitoksiinit ovat bakteerimyrkkyjä, joilla voi olla suuri vaikutus MS-taudin patogeneesiin.
PTX:llä voi olla erilaisia suojaavia vaikutuksia. EAE-hiirimallissa PTX vähensi demyelinaatiota jopa 75%. PTX vähentää myös lymfosyyttien tunkeutumista keskushermostoon, deaktivoi mikroglia-aktivaation ja muuttaa T-soluprofiilia lisäämällä T-auttajatyyppejä 1 ja 2 sekä T-säätelysoluja [84].
PTX-hoito saattaa suojata keskushermostoa autoimmuunisairaudelta säätelysytoksiinien säätelyn ja CD4 + CD25+ FoxP3+ Treg -solujen kautta. Bakteereista peräisin olevan toksiinin, hinkuyskätoksiinin, tiedetään alentavan herkkyyttä EAE:lle huolimatta siitä, että sen injektiota tarvitaan sairauden indusoimiseksi joissakin hiirikannoissa [85,86].
Botuliinitoksiini lamauttaa lihakset ja sitä käytetään perinteisenä spastisuuden hoitona [87]. On raportoitu, että tämä neurotoksiini saattaa parantaa monien MS-potilaiden elämänlaatua [88,89].
Tietyt patogeeniset sienet, jotka on erotettu ei-hermosolujen kudoksista, vapauttavat toksiineja, jotka kohdistuvat astrosyytteihin ja oligodendrosyytteihin aiheuttaen myeliinin hajoamista ja voivat laukaista MS-taudin [90]. Lisäksi erilaisten nekrotisoivien tekijöiden eritys aivojen aspergilloosissa voi aiheuttaa aivovaurioita ja vahingoittaa elintärkeitä soluja [91].
Ruokaan liittyvä mykotoksiini-okratoksiini A vaikuttaa haitallisesti lukuisiin solutyyppeihin, kuten astrosyytteihin [92]. Candida-infektioon havaittiin liittyvän lisääntynyt MS-kerroin [93]. Useat raportit osoittivat, että MS-potilailla voi olla vasta-aineita eri Candida-lajeja vastaan [94], mikä viittaa siihen, että tämä sieni-infektio voi olla MS-taudin riskitekijä [95]. Lisäksi C. Albicans -infektio ennen EAE-induktiota hiirillä pahentaa tautia. Samanlainen vaikutus on havaittu MS-potilailla [96].
Tarkastelemalla sienisoluseinän rakennetta, päällystävä liukenematon N-asetyyliglukosamiinipolymeeri hydrolysoidaan yleensä kitotriosidaasilla ’Chit’, joka on rakenteellisesti homologinen kitinaasien kanssa [97].
Aktivoidut makrofagit syntetisoivat ja erittävät kitinaaseja, jotka ovat kitiiniä hajottavia entsyymejä [98]. Sotgiu S et al. havaitsi, että mikrogliasta johdettu Chit-aktiivisuus MS-taudissa voi suojata aivoja kitiinimäisen aineen kerrostumiselta ja sen aiheuttamalta neurodegeneraatiolta [97]. Lisääntynyttä Chit-aktiivisuutta on havaittu erilaisia neurologisia häiriöitä sariastavavien potilaiden keskushermostossa [98] sekä MS-potilaiden plasmassa [99].
Tutkimuksissa on havaittu, että kitinaasien määrä on lisääntynyt neuromyeliittiä sairastavien potilaiden keskushermostossa vasteena IL-13:lle, mikä johtaa keskushermostotulehdukseen immuunisolujen lisääntyneiden kulkeutumisen veri-aivoesteen läpi [100] välityksellä.
Maantieteellisesti torajyväsienten esiintyminen osoitti merkittävää vastaavuutta MS-taudin maantieteellisen jakauman kanssa [101]. Sieni-infektio voi laukaista multippeliskleroosin tai se voi johtua MS-tautiin liittyvästä immuunijärjestelmän toimintahäiriöistä [102].
7. Kemikaalien, orgaanisten liuottimien ja raskasmetallien vaikutus
Monissa tutkimuksissa altistumista kemikaaleille, raskasmetalleille ja orgaanisille liuottimille pidetään potentiaalisina etiologisina tekijöinä, jotka myötävaikuttavat MS: n puhkeamiseen [103].
Esimerkiksi tinaa, hiilioksidia ja elohopeaa, mutta ei sinkkiä tai mangaania, pidetään MS-taudin riskiä lisäävinä ympäristötekijöinä [104]. Alueilla, joilla käytetään paljon kemikaaleja, kuten torjunta-aineita, MS-taudin esiintyvyysaste on ollut korkeampi [105,106,107,108]. Lisäksi torjunta-aineille altistuneilla maataloudessa työskentelevillä työntekijöillä oli suurempi riski sairastua MS-tautiin [109]. Tämä pätee erityisesti naisiin [110].
Ympäristön toksiinit voivat altistaa erityisesti odottavia naisia, mikä vaikuttaa sikiön kehitykseen ja syntyvän lapsen alttiuteen sairastua myöhemmin [111]. Kemikaaleille altistuneilla henkilöillä, kuten kenkä-, nahka- ja koneteollisuuden työntekijöillä, oli suurempi riski MS-taudin kehittymiseen [109, 112].
MS-taudin korkeampi esiintyvyys assosioituu alueisiin, jotka ovat erittäin saastuneita raskasmetalleista [113], kuten Isfahan, Iranin kolmanneksi suurin kaupunki [114] ja Lounais-Sardinia [115]. Raskasmetallien saannin ja neurodegeneratiivisten patologioiden välillä on myös dokumentoitu korrelaatio [116, 117].
Elohopean (Hg) on raportoitu liittyvän autoimmuniteettiin [118], koska se voi aiheuttaa oksidatiivista stressiä sekä vahingoittaa DNA:ta, mitokondrioita ja lipidikalvoja [119]. Lisäksi toistuva altistuminen elohopealle eläinkokeissa nopeutti taudin etenemistä mitokondrioiden aiheuttamien vaurioiden kautta [120].
Elohopea vaikuttaa astrosyytteihin, aivokuoren oligodendrosyytteihin, kortikomotoneuroneihin ja locus coeruleus -neuroneihin. Tämä saattaa selittää elohopean assosiaation MS-tautiin ja muihin keskushermoston degeneratiivisiin sairauksiin [119].
Useat raportit ovat osoittaneet, että seerumin hermospesifinen enolaasi (NSE-biomarkkeri elohopean neurotoksisille vaikutuksille) liittyy multippeliskleroosin etenemiseen [121]. Elohopeaa sisältävät hampaiden amalgaamitäytteet lisäsivät MS-taudin riskiä [122, 123]. Havainnoista huolimatta neurodegeneratiivisia sairauksia sairastavilta potilailta saatu tutkimusnäyttö osoitti vain epävarman yhteyden elohopean mahdollisesta osallisuudesta MS-taudin patogeneesiin [124]. Amalgaamipaikkoja ei juurikaan enää käytetä, joten amalgaami ei yksin selitä MS-tautiin sairastumista nykyään, vaikka se on saattanut olla yksi osatekijät niillä, joilla amalgaamipaikkoja on. Sen sijaan se saattaa pahentaa neurologisia oireita.
Toinen maaperässä esiintyvä myrkyllinen raskasmetalli on lyijy, joka näyttää lisäävän MS-taudin riskiä erityisesti miehillä [125]. Korkea lyijytoksisuus ja sen kyky säilyä ihmiskehossa pitkän aikaa tekevät siitä epäilyn riskitekijän monien selittämättömien sairauksien patogeneesissä [126].
MS-taudin riskin havaittiin kasvaneen 1,17 kertaa veren lyijypitoisuuden yhden ug / l lisäystä kohti [126]. Eräässä toisessa tutkimuksessa osoitettiin kuitenkin, että MS-tapaukset eivät näyttäneet keskittyvän lyijysulattojen ympärille [32].
Arseenia on myös maaperässä, ja altistuminen arseenille näyttää assosioituvan erityisesti naisten MS-tautiin sairastuvuuteen [125]. Arseeni voi aiheuttaa MS-taudin indusoimalla hermosolujen tulehdusreaktioita, rappeutumista ja apoptoosia, mukaan lukien hyperfosforylaatio ja tau-proteiinien aggregaatio, mikä johtaa tau-toiminnan deregulaatioon [127].
Sen sijaan kuparia käytetään myeliinin synteesissä, joten sen puute saattaa aiheuttaa myelopatiaa [128]. Muiden metallien vaikutuksista on kiistanalaista kirjallisuutta. MS-potilailla on todettu sinkin alhaisempia seerumitasoja [129], kun taas toinen tutkimus osoitti, että sinkkipitoisuudet lisääntyvät MS-potilailla. Nämä tulokset viittaavat siihen, että sinkin pitoisuuksien muutokset voivat olla osallisina MS:n patogeneesissä [130].
8. Lääkkeiden kehityksestä
Eräitä luonnollisia toksiineja on kaavailtu MS-taudin terapiavaihtoehdoiksi. Uusia yhdisteitä on eristetty niveljalkaisista ja muista myrkyllisistä eläimistä neurodegeneratiivisten sairauksien, kuten MS, hoitamiseksi [131].
Näihin kuuluvat ShK, merivuokon toksiini (Stichodactyla helianthus) ja skorpionimyrkkykomponentit, jotka ovat selektiivisiä kaliumkanavien salpaajia, joita tarvitaan aktivoituneiden T-lymfosyyttien toimintaan. Mehiläismyrkkyn (Apis mellifera) havaittiin myös parantavan taudin oireita, parantavan motorista toimintaa ja vähentävän tulehdusmerkkejä. Jopa käärmemyrkkyillä havaittiin olevan vaikutus MS-terapiassa, koska se estää autoimmuunisen enkefalomyeliitin ja lymfosyyttien aivojen tunkeutumisen kliinisiä oireita [132,133].
Uudet molekyylit, jotka on johdettu Thalassophryne nattereri Brasilian kalan myrkystä, ns. TnP-perhe, tuottavat systeemisiä ja keskushermostospesifisiä vaikutuksia, jotka estävät tulehduksellisten leukosyyttien migraation keskushermostoon ja demyelinaation ja voivat siten olla terapeuttinen vaihtoehto MS-taudin hoidossa [134].
Useat lääkkeet, kuten glatirameeriasetaatti, fingolimodi (FTY720), mitoksantroni, IFN-β, fumaarihappoesterit ja kortikosteroidit [2 135 136] voivat vähentää MS-taudin oireita.
Eräät lääkkeet vähentävät MS-riskiä, kuten tetanustoksoidirokotus, antibiootit, antihistamiinit ja sienilääkkeet. Niiden erityinen rooli on kuitenkin vielä puutteellisesti dokumentoitu [31]. Tetanustoksoidirokotusten raportoitiin vähentävän MS: n riskiä kolmanneksella rokotetuissa henkilöissä verrattuna rokottamatta jättämiseen [137]. Antibioottien suhteen kuvattiin korrelaatio penisilliinin käytön ja pienemmän multippeliskleroosiriskin välillä [138]. Muilla lääkkeillä, kuten antihistamiineilla, voi olla mahdollinen hyödyllinen vaikutus, jos niitä otetaan käyttöön MS-taudin puhkeamisen aikana [139], kun taas kolesterolia alentavien statiinien havaittiin vaikuttavan estävästi krooniseen ja uusiutuvaan EAE-malliin [140].
Kohtauksenvastainen valproiinihappo (VPA) suorittaa toimintansa lisäämällä asetyloituja histonitasoja, mikä johtaa lisääntyneeseen apoptoosiin neokorteksissa ja vähentyneeseen solujen lisääntymiseen ganglionisen eminenanssin yhteydessä [141]. Lisäksi VPA auttaa MS:n vaurioiden remyelinoinnissa lisäämällä endogeenisia esiasteita [142] ja voi vähentää selkäytimen tulehdusta aktivoituneissa T-soluissa tapahtuvan apoptoosin kautta [143]. Lisäksi VPA säätelee Th1- ja Th17-soluja alaspäin ja vähentää siten tulehduksellisia sytokiinitasoja [144].
Tulehduskipulääkkeiden havaittiin vaikuttavan immuunijärjestelmään, joten niillä voi olla terapeuttinen arvo MS-taudissa. P-amyriini, kannabinoidireseptorin agonisti, vähentää tulehdusta mikrogliaalisoluissa ja sitä voidaan käyttää potentiaalisena tulehdusta estävänä aineena keskushermostossa erityisesti neurodegeneratiivisissa sairauksissa. Tämä lääke vaikuttaa tulehdusvälittäjäprofiiliin vähentämällä TNF-alfaa, IL-1β: tä, IL-6: ta, PGE-2: ta, COX-2: ta sekä makrofagien M1 / M2-tasapainon säätelyä ja mikroglia-erilaistumista [145].
Toinen uusi aine on WWL70, anti-inflammatorinen terapeuttinen lääke, joka vaikuttaa mikrogliaan EAE-hiiren aivoissa vähentämällä COX-2:n ja mikrosomaalisen PGE2:n ilmentymistä [146]. Uusi yhdiste JC-171 (hydroksyylisulfonamidianalogi) toimii selektiivisenä NLRP3-tulehduksen estäjänä.
EAE-hiirimallissa JC-171: n raportoitiin estävän taudin etenemistä ja vakavuutta sekä ennalta ehkäisevissä että terapeuttisissa kokeellisissa järjestelyissä, mikä kannusti sen käyttöä MS-taudin terapiana [147]. Lisäksi securiniinilla, Securinega suffruticosa -kasvin juuresta peräisin olevalla tärkeimmällä luonnollisella alkaloidituotteella, on raportoitu olevan voimakas biologinen aktiivisuus estämällä merkittävästi NO-tuotantoa astrosyytteissä ja mikrogliassa sekä estämällä tulehduksellinen välittäjä NF-κB ja mitogeeni -aktivoidut proteiinikinaasit (MAPK). Siksi sitä voitaisiin käyttää potentiaalisena terapeuttisena kandidaattina neuroinflammaatioon liittyville sairauksille [148].
Fumaarihappoestereitä, kuten monometyylifumaraatti (MMF) ja dimetyylifumaraatti (DMF), on tutkittu intensiivisesti viime vuosina. DMF on hyväksytty erilaisten tulehdusvälitteisten sairauksien, mukaan lukien MS, hoitoon [149 150 151]. DMF vaikuttaa immuunijärjestelmän säätelyyn siirtymällä kohti Th2-sytokiiniprofiilia ja vähentämällä Th1- ja Th17-solujen vaikutusta. Vielä merkittävämmin DMF:llä ja sen metaboliitilla MMF:llä on antioksidanttinen ominaisuus aktivoimalla tumatekijä (erytroidista johdettu 2) kaltainen2 (NRF2), stimuloiden siten gliasolujen, oligodendrosyyttien ja neuronien sytosuojausta [152,153].
DMF:n on raportoitu vaikuttavan myeloidisoluihin sekä lymfosyytteihin, mukaan lukien B-solut ja luonnolliset tappajapopulaatiot (154, 155). Ryhmämme tutki DMF:n ja MMF:n vaikutusta NK-soluihin, missä havaitsimme, että ne lisäävät NK-kemotoksista ja sytolyyttistä toimintaa in vitro [155,156,157].
MMF parantaa EAE:n hiirillä aktivoimalla NK-soluja [42]. Fingolimodin (Gilenya), joka tunnetaan myös nimellä FTY720, synteettisen yhdisteen, joka jäljittelee sienen sekundääristä metaboliittia myriosiinia (ISP-I), ilmoitettiin olevan voimakas immunosuppressantti, jonka Yhdysvaltain FDA hyväksyi MS:n terapeuttisena aineena [158]. On arveltu, että FTY720 vaikuttaa immuunisolujen, kuten NK-solujen, aktiivisuuteen säätelemällä niiden aktivoimat reseptorit uudelleen ja tehostamalla niiden lyyttistä aktiivisuutta dendriittisoluja vastaan [159].
9. Päätelmä
Tässä katsauksessa avattiin alustavasti MS-tautiin vaikuttavien geneettisten ja epigeneettisten kemikaalien, toksiinien ja fysikaalisten tekijöiden monimuotoisuutta. Nämä tekijät voivat moduloida immuunijärjestelmän toimintaa epäsuorasti muuttamalla kehon autoantigeenejä, joita voidaan jakaa keskushermoston antigeenien kanssa, tai vapauttamalla immuunijärjestelmä reagoimatta estosignaaleihin. Epidemiologisten, kokeellisten tai kliinisten löydösten epäjohdonmukaisuus voi johtua paikallisista ja alueellisista vaihteluista sekä ympäristössä että väestögenetiikassa.
Lopuksi: Korrelaatio erilaisten toksiinien ja MS-taudin välillä on vahva. Sen sijaan kausaation osoittaminen on osoittautunut vaikeammaksi. Rokotteiden ja amalgaamipaikkojen raskasmetallit eivät selitä MS-tautiin sairastumista niillä, jotka eivät ole saaneet rokotusta tai joilla ei ole amalgaamipaikkoja. Luulen, että epigeneettisiä ympäristötekijöitä, jotka altistavat MS-taudille heikentämällä immuunijärjestelmää ja vaikuttamalla DNA:han on varmasti kymmenittäin.
Rokotteiden tai raskasmetallien vaikutusta MS-taudin laukaisijana ei voida täysin poissulkea, mutta riittävää näyttöä siitä, että ne aiheuttaisivat MS-taudin, ei kuitenkaan ole. Koronarokotteen suhteen olen luottavainen. Rokotteen aiheuttamat vakavat sivuoireet Yhdysvalloissa ja Britanniassa ovat olleet hyvin harvinaisia. Sen sijaan koronainfektio on aiheuttanut suurelle osalle sairastuneista pysyviä tai pitkäkestoisia oireita. Koska koronainfektion jälkitauteja ei tunneta, uskon, että rokottamisen riskit ovat merkittävästi pienemmät kuin taudin sairastamisen riskit. Pahoittelen, jos tekstiin jäi kirjoitus- ja käännösvirheitä.
References
- Mahad, D.H.; Trapp, B.D.; Lassmann, H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015, 14, 183–193. [Google Scholar] [CrossRef]
- De Souza, J.M.; Goncalves, B.D.C.; Gomez, M.V.; Vieira, L.B.; Ribeiro, F.M. Animal toxins as therapeutic tools to treat neurodegenerative diseases. Front. Pharmacol. 2018, 9, 145. [Google Scholar] [CrossRef] [PubMed]
- Zeller, D.; Classen, J. Plasticity of the motor system in multiple sclerosis. Neuroscience 2014, 283, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Browne, P.; Chandraratna, D.; Angood, C.; Tremlett, H.; Baker, C.; Taylor, B.V.; Thompson, A.J. Atlas of multiple sclerosis 2013: A growing global problem with widespread inequity. Neurology 2014, 83, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Dilokthornsakul, P.; Valuck, R.J.; Nair, K.V.; Corboy, J.R.; Allen, R.R.; Campbell, J.D. Multiple sclerosis prevalence in the united states commercially insured population. Neurology 2016, 86, 1014–1021. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, B.; Adamczyk-Sowa, M. New insights into the role of oxidative stress mechanisms in the pathophysiology and treatment of multiple sclerosis. Oxid. Med. Cell Longev. 2016, 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Bradl, M. Multiple sclerosis: Experimental models and reality. Acta Neuropathol. 2017, 133, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Comi, G.; Radaelli, M.; Soelberg Sørensen, P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet 2017, 389, 1347–1356. [Google Scholar] [CrossRef]
- Murray, T.J. The history of multiple sclerosis: The changing frame of the disease over the centuries. J. Neurol. Sci. 2009, 277, S3–S8. [Google Scholar] [CrossRef]
- Gu, C. Kir4.1: K(+) channel illusion or reality in the autoimmune pathogenesis of multiple sclerosis. Front. Mol. Neurosci. 2016, 9, 90. [Google Scholar] [CrossRef] [PubMed]
- Høglund, R.A.; Maghazachi, A.A. Multiple sclerosis and the role of immune cells. World J. Exp. Med. 2014, 4, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A.A. On the role of natural killer cells in neurodegenerative diseases. Toxins (Basel) 2013, 5, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.P.; Kermode, A.G.; Lucas, R.M.; Carroll, W.M.; Nolan, D.; Hart, P.H. Circulating immune cells in multiple sclerosis. Clin. Exp. Immunol. 2016, 187, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Sambucci, M.; Battistini, L.; Borsellino, G. Fas–fas ligand: Checkpoint of t cell functions in multiple sclerosis. Front. Immunol. 2016, 7, 382. [Google Scholar] [CrossRef] [PubMed]
- Elyaman, W.; Khoury, S.J. Th9 cells in the pathogenesis of eae and multiple sclerosis. Semin. Immunopathol. 2017, 39, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Fard, N.A.; Azizi, G.; Mirshafiey, A. The potential role of t helper cell 22 and il-22 in immunopathogenesis of multiple sclerosis. Innov. Clin. Neurosci. 2016, 13, 30–36. [Google Scholar] [PubMed]
- Hestvik, A.L.K. The double-edged sword of autoimmunity: Lessons from multiple sclerosis. Toxins (Basel) 2010, 2, 856–877. [Google Scholar] [CrossRef] [PubMed]
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, e00362. [Google Scholar] [CrossRef] [PubMed]
- Huynh, J.L.; Casaccia, P. Epigenetic mechanisms in multiple sclerosis: Implications for pathogenesis and treatment. Lancet Neurol. 2013, 12, 195–206. [Google Scholar] [CrossRef]
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2016, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Riemann-Lorenz, K.; Eilers, M.; von Geldern, G.; Schulz, K.-H.; Köpke, S.; Heesen, C. Dietary interventions in multiple sclerosis: Development and pilot-testing of an evidence based patient education program. PLoS ONE 2016, 11, e0165246. [Google Scholar] [CrossRef] [PubMed]
- Peedicayil, J. Epigenetic drugs for multiple sclerosis. Curr. Neuropharmacol. 2016, 14, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Babenko, O.; Kovalchuk, I.; Metz, G.A. Epigenetic programming of neurodegenerative diseases by an adverse environment. Brain Res. 2012, 1444, 96–111. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.W.; Metz, L.M.; Kovalchuk, O. Epigenetic changes in patients with multiple sclerosis. Nat. Rev. Neurol. 2012, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.W.; Alfredsson, L.; Costenbader, K.H.; Kamen, D.L.; Nelson, L.M.; Norris, J.M.; De Roos, A.J. Epidemiology of environmental exposures and human autoimmune diseases: Findings from a national institute of environmental health sciences expert panel workshop. J. Autoimmun. 2012, 39, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Alfredsson, L.; Olsson, T. Environmental factors and their interactions with risk genotypes in ms susceptibility. Curr. Opin. Neurol. 2016, 29, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Kakalacheva, K.; Lünemann, J.D. Environmental triggers of multiple sclerosis. FEBS Lett. 2011, 585, 3724–3729. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Loken-Amsrud, K.I.; Lossius, A.; Torkildsen, O.; Holmoy, T. Impact of the environment on multiple sclerosis. Tidsskr. Nor. Laegeforen. 2015, 135, 856–860. [Google Scholar] [PubMed]
- Milo, R.; Kahana, E. Multiple sclerosis: Geoepidemiology, genetics and the environment. Autoimmun. Rev. 2010, 9, A387–A394. [Google Scholar] [CrossRef] [PubMed]
- Turabelidze, G.; Schootman, M.; Zhu, B.P.; Malone, J.L.; Horowitz, S.; Weidinger, J.; Williamson, D.; Simoes, E. Multiple sclerosis prevalence and possible lead exposure. J. Neurol. Sci. 2008, 269, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Helmick, C.G.; Wrigley, J.M.; Zack, M.M.; Bigler, W.J.; Lehman, J.L.; Janssen, R.S.; Hartwig, E.C.; Witte, J.J. Multiple sclerosis in key west, florida. Am. J. Epidemiol. 1989, 130, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Forbes, R.B.; Wilson, S.V.; Swingler, R.J. The prevalence of multiple sclerosis in tayside, scotland: Do latitudinal gradients really exist? J. Neurol. 1999, 246, 1033–1040. [Google Scholar] [CrossRef] [PubMed]
- Gray, O.M.; McDonnell, G.V.; Hawkins, S.A. Factors in the rising prevalence of multiple sclerosis in the north-east of ireland. Mult. Scler. 2008, 14, 880–886. [Google Scholar] [CrossRef] [PubMed]
- Beck, C.A.; Metz, L.M.; Svenson, L.W.; Patten, S.B. Regional variation of multiple sclerosis prevalence in canada. Mult. Scler. 2005, 11, 516–519. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; McCarthy, A.; Quigley, C.; Bannan, L.; Hawkins, S.; Hutchinson, M. Latitudinal variation in the prevalence of multiple sclerosis in ireland, an effect of genetic diversity. J. Neurol. Neurosurg. Psychiatry 2004, 75, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.M.; Byrne, S.N.; Correale, J.; Ilschner, S.; Hart, P.H. Ultraviolet radiation, vitamin d and multiple sclerosis. Neurodegener. Dis. Manag. 2015, 5, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, F.M. Update in vitamin d and multiple sclerosis. Neurosciences (Riyadh) 2015, 20, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Niino, M.; Sato, S.; Fukazawa, T.; Masaki, K.; Miyazaki, Y.; Matsuse, D.; Yamasaki, R.; Takahashi, E.; Kikuchi, S.; Kira, J. Decreased serum vitamin d levels in japanese patients with multiple sclerosis. J. Neuroimmunol. 2015, 279, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Munger, K.L.; Zhang, S.M.; O’Reilly, E.; Hernan, M.A.; Olek, M.J.; Willett, W.C.; Ascherio, A. Vitamin d intake and incidence of multiple sclerosis. Neurology 2004, 62, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Vitamin d3 and monomethyl fumarate enhance natural killer cell lysis of dendritic cells and ameliorate the clinical score in mice suffering from experimental autoimmune encephalomyelitis. Toxins (Basel) 2015, 7, 4730–4744. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, N.M.; Munger, K.L.; Koch-Henriksen, N.; Hougaard, D.M.; Magyari, M.; Jorgensen, K.T.; Lundqvist, M.; Simonsen, J.; Jess, T.; Cohen, A.; et al. Neonatal vitamin d status and risk of multiple sclerosis: A population-based case-control study. Neurology 2017, 88, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Fernandes de Abreu, D.A.; Babron, M.C.; Rebeix, I.; Fontenille, C.; Yaouanq, J.; Brassat, D.; Fontaine, B.; Clerget-Darpoux, F.; Jehan, F.; Feron, F. Season of birth and not vitamin d receptor promoter polymorphisms is a risk factor for multiple sclerosis. Mult. Scler. 2009, 15, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Saccone, D.; Asani, F.; Bornman, L. Regulation of the vitamin d receptor gene by environment, genetics and epigenetics. Gene 2015, 561, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, A.O.; Sanseverino, I.; Purificato, C.; Cortese, A.; Mechelli, R.; Francisci, S.; Salvetti, M.; Millefiorini, E.; Gessani, S.; Gauzzi, M.C. Increased circulating levels of vitamin d binding protein in ms patients. Toxins (Basel) 2015, 7, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Hutter, C.D.; Laing, P. Multiple sclerosis: Sunlight, diet, immunology and aetiology. Med. Hypotheses 1996, 46, 67–74. [Google Scholar] [PubMed]
- Hedstrom, A.K.; Akerstedt, T.; Hillert, J.; Olsson, T.; Alfredsson, L. Shift work at young age is associated with increased risk for multiple sclerosis. Ann. Neurol. 2011, 70, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Ponsonby, A.-L.; Lucas, R.M. Shift work and multiple sclerosis. Ann. Neurol. 2011, 70, 680–683. [Google Scholar] [CrossRef] [PubMed]
- Gustavsen, S.; Sondergaard, H.B.; Oturai, D.B.; Laursen, B.; Laursen, J.H.; Magyari, M.; Ullum, H.; Larsen, M.H.; Sellebjerg, F.; Oturai, A.B. Shift work at young age is associated with increased risk of multiple sclerosis in a danish population. Mult. Scler. Relat. Disord. 2016, 9, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Akerstedt, T.; Olsson, T.; Alfredsson, L. Shift work influences multiple sclerosis risk. Mult. Scler. 2015, 21, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, R.; Li, Z.; Wang, Y.; Gao, C.; Lv, X.; Song, Y.; Li, B. The risk of smoking on multiple sclerosis: A meta-analysis based on 20,626 cases from case-control and cohort studies. PeerJ 2016, 4, e1797. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.P.; Hartoonian, N.; Maynard, C.; Leipertz, S.L.; Haselkorn, J.K. Smoking and physical activity: Examining health behaviors and 15-year mortality among individuals with multiple sclerosis. Arch. Phys. Med. Rehabil. 2015, 96, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Ramanujam, R.; Hedström, A.; Manouchehrinia, A.; Alfredsson, L.; Olsson, T.; Bottai, M.; Hillert, J. Effect of smoking cessation on multiple sclerosis prognosis. JAMA Neurol. 2015, 72, 1117–1123. [Google Scholar] [CrossRef] [PubMed]
- Öckinger, J.; Hagemann-Jensen, M.; Kullberg, S.; Engvall, B.; Eklund, A.; Grunewald, J.; Piehl, F.; Olsson, T.; Wahlström, J. T-cell activation and hla-regulated response to smoking in the deep airways of patients with multiple sclerosis. Clin. Immunol. 2016, 169, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part ii: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Bomfim, I.L.; Barcellos, L.F.; Briggs, F.; Schaefer, C.; Kockum, I.; Olsson, T.; Alfredsson, L. Interaction between passive smoking and two hla genes with regard to multiple sclerosis risk. Int. J. Epidemiol. 2014, 43, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Timmermans, S.; Bogie, J.F.J.; Vanmierlo, T.; Lütjohann, D.; Stinissen, P.; Hellings, N.; Hendriks, J.J.A. High fat diet exacerbates neuroinflammation in an animal model of multiple sclerosis by activation of the renin angiotensin system. J. Neuroimmune Pharmacol. 2014, 9, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Farinotti, M.; Vacchi, L.; Simi, S.; Di Pietrantonj, C.; Brait, L.; Filippini, G. Dietary interventions for multiple sclerosis. Cochrane Database Syst. Rev. 2012, 12, CD004192. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.-D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Mowry, E.M.; Gianfrancesco, M.A.; Shao, X.; Schaefer, C.A.; Shen, L.; Olsson, T.; Barcellos, L.F.; Alfredsson, L. High consumption of coffee is associated with decreased multiple sclerosis risk; results from two independent studies. J. Neurol. Neurosurg. Psychiatry 2016, 87, 454–460. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mowry, E.; Hedstrom, A.; Gianfrancesco, M.; Shao, X.; Schaefer, C.; Barcellos, L.; Olsson, T.; Alfredsson, L. Greater consumption of coffee is associated with reduced odds of multiple sclerosis (s45.004). Neurology 2015, 84, S45.004. [Google Scholar]
- D’hooghe, M.B.; Haentjens, P.; Nagels, G.; De Keyser, J. Alcohol, coffee, fish, smoking and disease progression in multiple sclerosis. Eur. J. Neurol. 2011, 19, 616–624. [Google Scholar] [CrossRef] [PubMed]
- Farez, M.F.; Fiol, M.P.; Gaitán, M.I.; Quintana, F.J.; Correale, J. Sodium intake is associated with increased disease activity in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 2015, 86, 26. [Google Scholar] [CrossRef] [PubMed]
- Riccio, P.; Rossano, R. Nutrition facts in multiple sclerosis. ASN Neuro 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Mielcarz, D.W.; Kasper, L.H. The gut microbiome in multiple sclerosis. Curr. Treat. Opt. Neurol. 2015, 17, 18. [Google Scholar] [CrossRef] [PubMed]
- Kirby, T.O.; Ochoa-Repáraz, J. The gut microbiome in multiple sclerosis: A potential therapeutic avenue. Med. Sci. (Basel, Switzerland) 2018, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Lang, Y.; Shen, D.; Jin, T.; Zhu, J.; Cui, L. Gut microbiota in multiple sclerosis and experimental autoimmune encephalomyelitis: Current applications and future perspectives. Med. Inflamm. 2018, 2018, 8168717. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roy Sarkar, S.; Banerjee, S. Gut microbiota in neurodegenerative disorders. J. Neuroimmunol. 2019, 328, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Yokote, H.; Miyake, S.; Croxford, J.L.; Oki, S.; Mizusawa, H.; Yamamura, T. Nkt cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am. J. Pathol. 2008, 173, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Brewer, J.H.; Thrasher, J.D.; Straus, D.C.; Madison, R.A.; Hooper, D. Detection of mycotoxins in patients with chronic fatigue syndrome. Toxins (Basel) 2013, 5, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Ordog, G. 476 multiple sclerosis cluster: Mycotoxic leukoencephalopathy. J. Investig. Med. 2005, 53, S161. [Google Scholar] [CrossRef]
- Venkatesan, A. Multiple sclerosis and infections. Neurodegener. Dis. Manag. 2015, 5, 11–14. [Google Scholar] [CrossRef] [PubMed]
- Oleszak, E.L.; Chang, J.R.; Friedman, H.; Katsetos, C.D.; Platsoucas, C.D. Theiler’s virus infection: A model for multiple sclerosis. Clin. Microbiol. Rev. 2004, 17, 174–207. [Google Scholar] [CrossRef] [PubMed]
- Fong, I.W. The Role of Microbes in Common Non-Infectious Diseases; Springer: New York, NY, USA, 2014. [Google Scholar]
- Burns, J.; Littlefield, K.; Gill, J.; Trotter, J.L. Bacterial toxin superantigens activate human t lymphocytes reactive with myelin autoantigens. Ann. Neurol. 1992, 32, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Gay, F. Bacterial toxins and multiple sclerosis. J. Neurol. Sci. 2007, 262, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Blanch, M.; Dorca-Arévalo, J.; Not, A.; Cases, M.; Gómez de Aranda, I.; Martínez-Yélamos, A.; Martínez-Yélamos, S.; Solsona, C.; Blasi, J. The cytotoxicity of epsilon toxin from clostridium perfringens on lymphocytes is mediated by mal protein expression. Mol. Cell. Biol. 2018, 38, e00086-18. [Google Scholar] [CrossRef] [PubMed]
- Cases, M.; Llobet, A.; Terni, B.; Gómez de Aranda, I.; Blanch, M.; Doohan, B.; Revill, A.; Brown, A.M.; Blasi, J.; Solsona, C. Acute effect of pore-forming clostridium perfringens ε-toxin on compound action potentials of optic nerve of mouse. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Navarro, M.A.; Li, J.; Freedman, J.C.; Shrestha, A.; McClane, B.A. Comparative pathogenesis of enteric clostridial infections in humans and animals. Anaerobe 2018, 53, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens epsilon toxin causes selective death of mature oligodendrocytes and central nervous system demyelination. mBio 2015, 6, e02513–e02514. [Google Scholar] [CrossRef] [PubMed]
- Wagley, S.; Bokori-Brown, M.; Morcrette, H.; Malaspina, A.; D’Arcy, C.; Gnanapavan, S.; Lewis, N.; Popoff, M.R.; Raciborska, D.; Nicholas, R.; et al. Evidence of clostridium perfringens epsilon toxin associated with multiple sclerosis. Mult. Scler. J. 2018, 1352458518767327. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.-X.; Tang, Z.; Gan, Y.; Li, L.; Shi, F.; Coons, S.; Shi, J. Pertussis toxin modulates microglia and t cell profile to protect experimental autoimmune encephalomyelitis. Neuropharmacology 2014, 81, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.S.; Benkhoucha, M.; Lehmann-Horn, K.; Hertzenberg, D.; Sellner, J.; Santiago-Raber, M.-L.; Chofflon, M.; Hemmer, B.; Zamvil, S.S.; Lalive, P.H. Repetitive pertussis toxin promotes development of regulatory t cells and prevents central nervous system autoimmune disease. PLoS ONE 2011, 5, e16009. [Google Scholar] [CrossRef] [PubMed]
- Steelman, A.J. Infection as an environmental trigger of multiple sclerosis disease exacerbation. Front. Immunol. 2015, 6, 520. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D.; Bhidayasiri, R.; Bohlega, S.; Chahidi, A.; Chung, T.M.; Ebke, M.; Jacinto, L.J.; Kaji, R.; Koçer, S.; Kanovsky, P.; et al. Botulinum toxin therapy for treatment of spasticity in multiple sclerosis: Review and recommendations of the iab-interdisciplinary working group for movement disorders task force. J. Neurol. 2017, 264, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Cameron, M.H.; Bethoux, F.; Davis, N.; Frederick, M. Botulinum toxin for symptomatic therapy in multiple sclerosis. Curr. Neurol. Neurosci. Rep. 2014, 14, 463. [Google Scholar] [CrossRef] [PubMed]
- Latino, P.; Castelli, L.; Prosperini, L.; Marchetti, M.R.; Pozzilli, C.; Giovannelli, M. Determinants of botulinum toxin discontinuation in multiple sclerosis: A retrospective study. Neurol. Sci. 2017, 38, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Purzycki, C.B.; Shain, D.H. Fungal toxins and multiple sclerosis: A compelling connection. Brain Res. Bull. 2010, 82, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Speth, C.; Rambach, G.; Lass-Flörl, C.; Würzner, R.; Gasque, P.; Mohsenipour, I.; Dierich, M.P. Culture supernatants of patient-derived aspergillus isolates have toxic and lytic activity towards neurons and glial cells. FEMS Immunol. Med. Microbiol. 2006, 29, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Razafimanjato, H.; Garmy, N.; Guo, X.-J.; Varini, K.; Di Scala, C.; Di Pasquale, E.; Taïeb, N.; Maresca, M. The food-associated fungal neurotoxin ochratoxin a inhibits the absorption of glutamate by astrocytes through a decrease in cell surface expression of the excitatory amino-acid transporters glast and glt-1. Neurotoxicology 2010, 31, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Benito-León, J.; Pisa, D.; Alonso, R.; Calleja, P.; Díaz-Sánchez, M.; Carrasco, L. Association between multiple sclerosis and candida species: Evidence from a case-control study. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Carrasco, L. Fungal infection in a patient with multiple sclerosis. Eur. J. Clin. Microbiol. Infect. Dis. 2011, 30, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Pisa, D.; Alonso, R.; Jiménez-Jiménez, F.J.; Carrasco, L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur. J. Clin. Microbiol. Infect. Dis. 2013, 32, 795–801. [Google Scholar] [CrossRef] [PubMed]
- Fraga-Silva, T.F.C.; Mimura, L.A.N.; Marchetti, C.M.; Chiuso-Minicucci, F.; França, T.G.D.; Zorzella-Pezavento, S.F.G.; Venturini, J.; Arruda, M.S.P.; et al. Experimental autoimmune encephalomyelitis development is aggravated by candida albicans infection. J. Immunol. Res. 2015, 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Sotgiu, S.; Musumeci, S.; Marconi, S.; Gini, B.; Bonetti, B. Different content of chitin-like polysaccharides in multiple sclerosis and alzheimer’s disease brains. J. Neuroimmunol. 2008, 197, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Barone, R.; Sotgiu, S.; Musumeci, S. Plasma chitotriosidase in health and pathology. Clin. Lab. 2007, 53, 321–333. [Google Scholar] [PubMed]
- Comabella, M.; Domínguez, C.; Rio, J.; Martín-Gallán, P.; Vilches, A.; Vilarrasa, N.; Espejo, C.; Montalban, X. Plasma chitotriosidase activity in multiple sclerosis. Clin. Immunol. 2009, 131, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Correale, J.; Fiol, M. Chitinase effects on immune cell response in neuromyelitis optica and multiple sclerosis. Mult. Scler. J. 2010, 17, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, M. Multiple sclerosis—Is research on the wrong track? Med. Hypotheses 1991, 34, 69–72. [Google Scholar] [CrossRef]
- Ramos, M.; Pisa, D.; Molina, S.; Rábano, A.; Juarranz, A.; Carrasco, L. Fungal infection in patients with multiple sclerosis. Open Mycol. J. 2008, 2, 22–28. [Google Scholar] [CrossRef]
- Napier, M.D.; Poole, C.; Satten, G.A.; Ashley-Koch, A.; Marrie, R.A.; Williamson, D.M. Heavy metals, organic solvents and multiple sclerosis: An exploratory look at gene-environment interactions. Arch. Environ. Occup. Health 2016, 71, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Compston, A.; Lassmann, H.; McDonald, I.; Miller, D.; Noseworthy, J.; Smith, K.; Wekerle, H.; Confavreux, C. The story of multiple sclerosis. In Mcalpine’s Multiple Sclerosis, 4th ed.; Churchill Livingstone: London, UK, 2005. [Google Scholar]
- Parron, T.; Requena, M.; Hernandez, A.F.; Alarcon, R. Association between environmental exposure to pesticides and neurodegenerative diseases. Toxicol. Appl. Pharmacol. 2011, 256, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Savage, E.P.; Keefe, T.J.; Mounce, L.M.; Heaton, R.K.; Lewis, J.A.; Burcar, P.J. Chronic neurological sequelae of acute organophosphate pesticide poisoning. Arch. Environ. Health 1988, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Prou, D.; Perier, C.; Przedborski, S. Toxin-induced models of parkinson’s disease. NeuroRx 2005, 2, 484–494. [Google Scholar] [CrossRef] [PubMed]
- Dubey, D.; Sharma, V.; Stuve, O. Multiple mothballs or multiple sclerosis: A diagnostic dilemma (p5.192). Neurology 2014, 82, 192. [Google Scholar]
- Oddone, E.; Crosignani, P.; Scaburri, A.; Bai, E.; Modonesi, C.; Imbriani, M.; Bergamaschi, R. Occupation and multiple sclerosis: An italian case-control study. Occup. Environ. Med. 2013, 70, A91. [Google Scholar] [CrossRef]
- Magyari, M.; Koch-Henriksen, N.; Pfleger, C.C.; Sorensen, P.S. Physical and social environment and the risk of multiple sclerosis. Mult. Scler. Relat. Disord. 2014, 3, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.; Chitnis, T.; Weinstock-Guttman, B.; Rubin, J.; Zelikovitch, A.; Nourbakhsh, B.; Simmons, T.; Casper, C.; Waubant, E. Maternal illness in pregnancy and perinatal exposure to pesticides are associated with risk for pediatric onset ms (s29.005). Neurology 2016, 86, S29.005. [Google Scholar]
- Landtblom, A.-M.; Flodin, U.; Söderfeldt, B.; Wolfson, C.; Axelson, O. Organic solvents and multiple sclerosis: A synthesis of the current evidence. Epidemiology 1996, 7, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Iranmanesh, F.; Ebrahimi, H.a.; Iranmanesh, M.; Sedighi, B.; Gadari, F. Multiple sclerosis and mines: An epidemiologic study from kerman province, Iran. Int. Clin. Neurosci. J. 2015, 2, 133–136. [Google Scholar]
- Razavi, Z.; Jokar, M.; Allafchian, A.; Hossinpour, Z.; Berenjani, L.; Shayegan Nejad, V. The relationship between blood lead levels and clinical features among multiple sclerosis patients in Isfahan, Iran. Iran. J. Health, Saf. Environ. 2016, 3, 412–420. [Google Scholar]
- Monti, M.C.; Guido, D.; Montomoli, C.; Sardu, C.; Sanna, A.; Pretti, S.; Lorefice, L.; Marrosu, M.G.; Valera, P.; Cocco, E. Is geo-environmental exposure a risk factor for multiple sclerosis? A population-based cross-sectional study in south-western sardinia. PLoS ONE 2016, 11, e0163313. [Google Scholar] [CrossRef] [PubMed]
- Giacoppo, S.; Galuppo, M.; Calabro, R.S.; D’Aleo, G.; Marra, A.; Sessa, E.; Bua, D.G.; Potorti, A.G.; Dugo, G.; Bramanti, P.; et al. Heavy metals and neurodegenerative diseases: An observational study. Biol. Trace Elem. Res. 2014, 161, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Fulgenzi, A.; Zanella, S.G.; Mariani, M.M.; Vietti, D.; Ferrero, M.E. A case of multiple sclerosis improvement following removal of heavy metal intoxication: Lessons learnt from matteo’s case. Biometals 2012, 25, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Crowe, W.; Allsopp, P.J.; Watson, G.E.; Magee, P.J.; Strain, J.J.; Armstrong, D.J.; Ball, E.; McSorley, E.M. Mercury as an environmental stimulus in the development of autoimmunity—A systematic review. Autoimmun. Rev. 2017, 16, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Pamphlett, R.; Kum Jew, S. Inorganic mercury in human astrocytes, oligodendrocytes, corticomotoneurons and the locus ceruleus: Implications for multiple sclerosis, neurodegenerative disorders and gliomas. Biometals 2018, 31, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Pourahmad, J.; Kahrizi, F.; Naderi, N.; Salimi, A.; Noorbakhsh, F.; Faizi, M.; Naserzadeh, P. Repeated administration of mercury accelerates progression of multiple sclerosis through mitochondrial dysfunction. Iran. J. Pharm. Res. 2016, 15, 834–841. [Google Scholar]
- Guzzi, G.; Costa, A.; Pigatto, P. Serum nse and multiple sclerosis. J. Neurol. Sci. 2015, 358, 463. [Google Scholar] [CrossRef] [PubMed]
- Soni, R.; Bhatnagar, A.; Vivek, R.; Chaturvedi, T.; Singh, A. A systematic review on mercury toxicity from dental amalgam fillings and its management strategies. J. Sci. Res. 2012, 56, 81–92. [Google Scholar]
- Bjørklund, G.; Hilt, B.; Dadar, M.; Lindh, U.; Aaseth, J. Neurotoxic effects of mercury exposure in dental personnel. Basic Clin. Pharmacol. Toxicol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cariccio, V.L.; Samà, A.; Bramanti, P.; Mazzon, E. Mercury involvement in neuronal damage and in neurodegenerative diseases. Biol. Trace Elem. Res. 2019, 187, 341–356. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-P.; Lee, C.T.-C. Multiple sclerosis incidence associated with the soil lead and arsenic concentrations in taiwan. PLoS ONE 2013, 8, e65911. [Google Scholar] [CrossRef] [PubMed]
- Dehghanifiroozabadi, M.; Noferesti, P.; Amirabadizadeh, A.; Nakhaee, S.; Aaseth, J.; Noorbakhsh, F.; Mehrpour, O. Blood lead levels and multiple sclerosis: A case-control study. Mult. Scler. Relat. Disord. 2019, 27, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh-Ghodsi, M.; Zavvari, A.; Ebrahimi-Kalan, A.; Shiri-Shahsavar, M.R.; Yousefi, B. The hypothetical roles of arsenic in multiple sclerosis by induction of inflammation and aggregation of tau protein: A commentary. Nutr. Neurosci. 2018, 21, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Jaiser, S.R.; Winston, G.P. Copper deficiency myelopathy. J. Neurol. 2010, 257, 869–881. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Palm, R.; Hallmans, G. Zinc and copper in multiple sclerosis. J. Neurol. Neurosurg. Psychiatry 1982, 45, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Bredholt, M.; Frederiksen, J.L. Zinc in multiple sclerosis: A systematic review and meta-analysis. ASN Neuro 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Zahid Rasul, N.; Naqab, K.; Samiullah, K.; Mehboob, A.; Mohammad Amjad, K. Potential application of venom proteins in designing of medicines for treating human neurodegenerative disorders. Protein Pept. Lett. 2018, 25, 633–642. [Google Scholar]
- Iwai, S.; Okazaki, M.; Kiuchi, Y.; Oguchi, K. Changes in mrna levels of fibrinogen subunit polypeptides in rats defibrinogenated with batroxobin. Thromb. Res. 1999, 96, 421–426. [Google Scholar] [CrossRef]
- Hinman, C.L.; Stevens-Truss, R.; Schwarz, C.; Hudson, R.A. Sequence determinants of modified cobra venom neurotoxin which induce immune resistance to experimental allergic encephalomyelitis: Molecular mechans for immunologic action. Immunopharmacol. Immunotoxicol. 1999, 21, 483–506. [Google Scholar] [CrossRef] [PubMed]
- Komegae, E.N.; Souza, T.A.M.; Grund, L.Z.; Lima, C.; Lopes-Ferreira, M. Multiple functional therapeutic effects of tnp: A small stable synthetic peptide derived from fish venom in a mouse model of multiple sclerosis. PLoS ONE 2017, 12, e0171796. [Google Scholar] [CrossRef] [PubMed]
- Ontaneda, D.; Hyland, M.; Cohen, J.A. Multiple sclerosis: New insights in pathogenesis and novel therapeutics. Annu. Rev. Med. 2012, 63, 389–404. [Google Scholar] [CrossRef] [PubMed]
- Kamm, C.P.; Uitdehaag, B.M.; Polman, C.H. Multiple sclerosis: Current knowledge and future outlook. Eur. Neurol. 2014, 72, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Hernan, M.A.; Alonso, A.; Hernandez-Diaz, S. Tetanus vaccination and risk of multiple sclerosis: A systematic review. Neurology 2006, 67, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Jick, S.S.; Jick, H.; Hernan, M.A. Antibiotic use and risk of multiple sclerosis. Am. J. Epidemiol. 2006, 163, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Jick, S.S.; Hernan, M.A. Allergy, histamine 1 receptor blockers, and the risk of multiple sclerosis. Neurology 2006, 66, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Ifergan, I.; Wosik, K.; Cayrol, R.; Kébir, H.; Auger, C.; Bernard, M.; Bouthillier, A.; Moumdjian, R.; Duquette, P.; Prat, A. Statins reduce human blood-brain barrier permeability and restrict leukocyte migration: Relevance to multiple sclerosis. Ann. Neurol. 2006, 60, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Mabunga, D.F.N.; Gonzales, E.L.T.; Kim, J.-w.; Kim, K.C.; Shin, C.Y. Exploring the validity of valproic acid animal model of autism. Exp. Neurobiol. 2015, 24, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Pazhoohan, S.; Satarian, L.; Asghari, A.A.; Salimi, M.; Kiani, S.; Mani, A.R.; Javan, M. Valproic acid attenuates disease symptoms and increases endogenous myelin repair by recruiting neural stem cells and oligodendrocyte progenitors in experimental autoimmune encephalomyelitis. Neurodegener. Dis. 2014, 13, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Du, C.; Wei, W.; Wu, Z.; Zhao, G.; Li, Z.; Xie, X. The antiepileptic drug valproic acid restores t cell homeostasis and ameliorates pathogenesis of experimental autoimmune encephalomyelitis. J. Biol. Chem. 2012, 287, 28656–28665. [Google Scholar] [CrossRef] [PubMed]
- Long, J.; Chang, L.; Shen, Y.; Gao, W.H.; Wu, Y.N.; Dou, H.B.; Huang, M.M.; Wang, Y.; Fang, W.Y.; Shan, J.H.; et al. Valproic acid ameliorates graft-versus-host disease by downregulating th1 and th17 cells. J. Immunol. 2015, 195, 1849–1857. [Google Scholar] [CrossRef] [PubMed]
- Askari, V.R.; Fereydouni, N.; Baradaran Rahimi, V.; Askari, N.; Sahebkar, A.H.; Rahmanian-Devin, P.; Samzadeh-Kermani, A. B-amyrin, the cannabinoid receptors agonist, abrogates mice brain microglial cells inflammation induced by lipopolysaccharide/interferon-γ and regulates mφ1/mφ2 balances. Biomed. Pharmacother. 2018, 101, 438–446. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Moran, S.; Wen, J.; Affram, K.; Chen, T.; Symes, A.J.; Zhang, Y. Wwl70 attenuates pge(2) production derived from 2-arachidonoylglycerol in microglia by abhd6-independent mechanism. J. Neuroinflamm. 2017, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Fulp, J.W.; Jiang, Y.; Li, X.; Chojnacki, J.E.; Wu, J.; Wang, X.-Y.; Zhang, S. Development and characterization of a hydroxyl-sulfonamide analogue, 5-chloro-n-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel nlrp3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem. Neurosci. 2017, 8, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Leonoudakis, D.; Rane, A.; Angeli, S.; Lithgow, G.J.; Andersen, J.K.; Chinta, S.J. Anti-inflammatory and neuroprotective role of natural product securinine in activated glial cells: Implications for parkinson’s disease. Med. Inflamm. 2017, 2017, 8302636. [Google Scholar] [CrossRef] [PubMed]
- Kappos, L.; Gold, R.; Miller, D.H.; MacManus, D.G.; Havrdova, E.; Limmroth, V.; Polman, C.H.; Schmierer, K.; Yousry, T.A.; Yang, M.; et al. Efficacy and safety of oral fumarate in patients with relapsing-remitting multiple sclerosis: A multicentre, randomised, double-blind, placebo-controlled phase iib study. Lancet 2008, 372, 1463–1472. [Google Scholar] [CrossRef]
- Stangel, M.; Linker, R.A. Dimethyl fumarate (bg-12) for the treatment of multiple sclerosis. Expert Rev. Clin. Pharmacol. 2013, 6, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Gold, R. Dimethyl fumarate for treatment of multiple sclerosis: Mechanism of action, effectiveness, and side effects. Curr. Neurol. Neurosci. Rep. 2013, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Bomprezzi, R. Dimethyl fumarate in the treatment of relapsing–remitting multiple sclerosis: An overview. Ther. Adv. Neurol. Disord. 2015, 8, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Gopal, S.; Mikulskis, A.; Gold, R.; Fox, R.J.; Dawson, K.T.; Amaravadi, L. Evidence of activation of the nrf2 pathway in multiple sclerosis patients treated with delayed-release dimethyl fumarate in the phase 3 define and confirm studies. Mult. Scler. 2017, 23, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.A.; Ogrodnik, M.A.; Plave, A.; Mao-Draayer, Y. Emerging understanding of the mechanism of action for dimethyl fumarate in the treatment of multiple sclerosis. Front. Neurol. 2018, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Vego, H.; Sand, K.L.; Høglund, R.A.; Fallang, L.-E.; Gundersen, G.; Holmøy, T.; Maghazachi, A.A. Monomethyl fumarate augments nk cell lysis of tumor cells through degranulation and the upregulation of nkp46 and cd107a. Cell Mol. Immunol. 2014, 13, 57. [Google Scholar] [CrossRef] [PubMed]
- Maghazachi, A.A.; Sand, K.L.; Al-Jaderi, Z. Glatiramer acetate, dimethyl fumarate, and monomethyl fumarate upregulate the expression of ccr10 on the surface of natural killer cells and enhance their chemotaxis and cytotoxicity. Front. Immunol. 2016, 7, 437. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Utilization of dimethyl fumarate and related molecules for treatment of multiple sclerosis, cancer, and other diseases. Front. Immunol. 2016, 7, 278. [Google Scholar] [CrossRef] [PubMed]
- Strader, C.R.; Pearce, C.J.; Oberlies, N.H. Fingolimod (fty720): A recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod. 2011, 74, 900–907. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Effects of vitamin d3, calcipotriol and fty720 on the expression of surface molecules and cytolytic activities of human natural killer cells and dendritic cells. Toxins (Basel) 2013, 5, 1932–1947. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).































 Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/
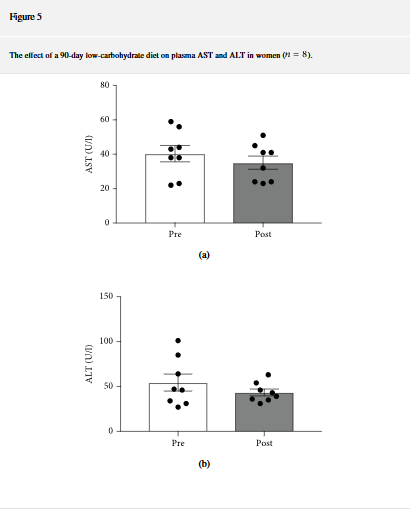
Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/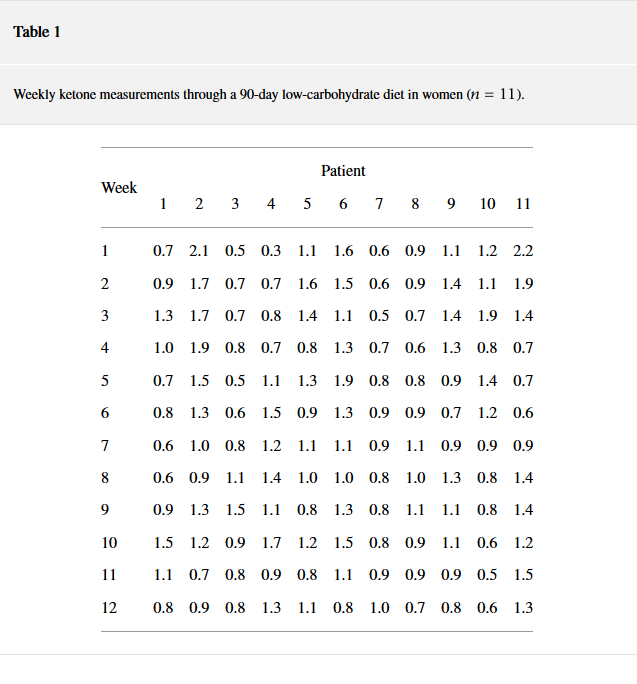






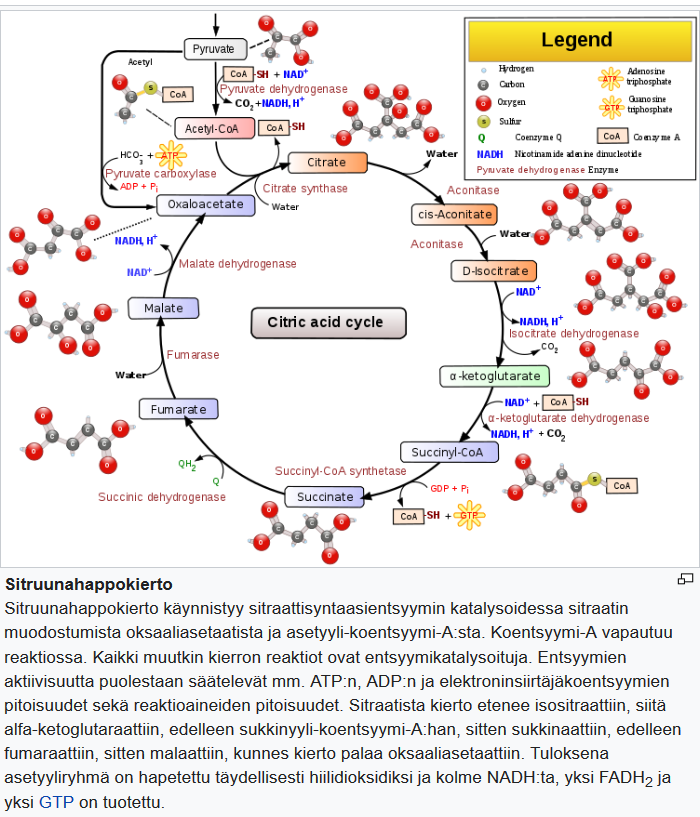
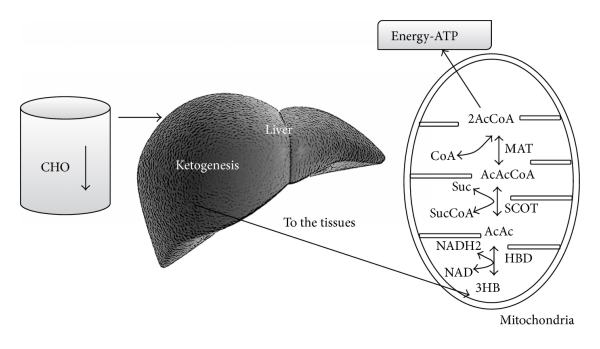 Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.
Keskushermostossa glukoosia tarvitaan energia lähteeksi, sekä tuottamaan pyruvaatteja, jotka voidaan edelleen muuntaa oksaloasetaatiksi.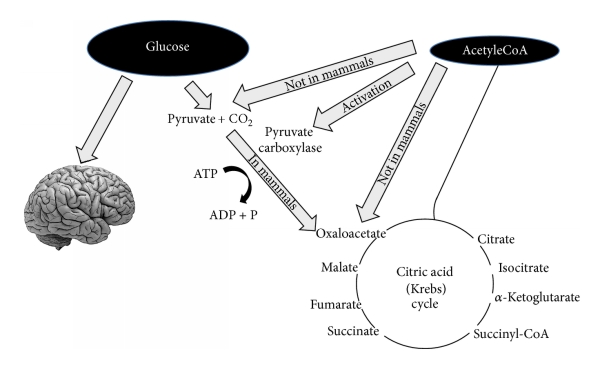














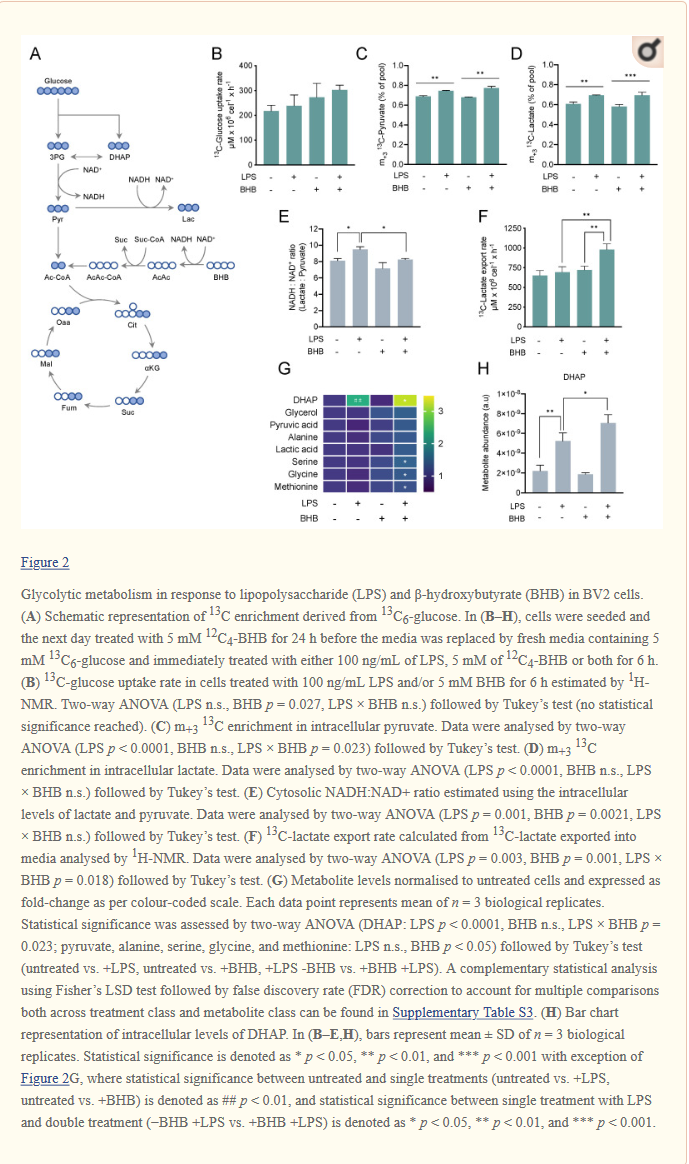







 C-peptidi muodostuu haiman Langerhansin saarekkeiden β-soluissa syntetisoidusta proinsuliinista sen pilkkoutuessa insuliiniksi ja C-peptidiksi. Sitä erittyy vereen insuliinin kanssa ekvimolaarisina määrinä.
C-peptidi muodostuu haiman Langerhansin saarekkeiden β-soluissa syntetisoidusta proinsuliinista sen pilkkoutuessa insuliiniksi ja C-peptidiksi. Sitä erittyy vereen insuliinin kanssa ekvimolaarisina määrinä.