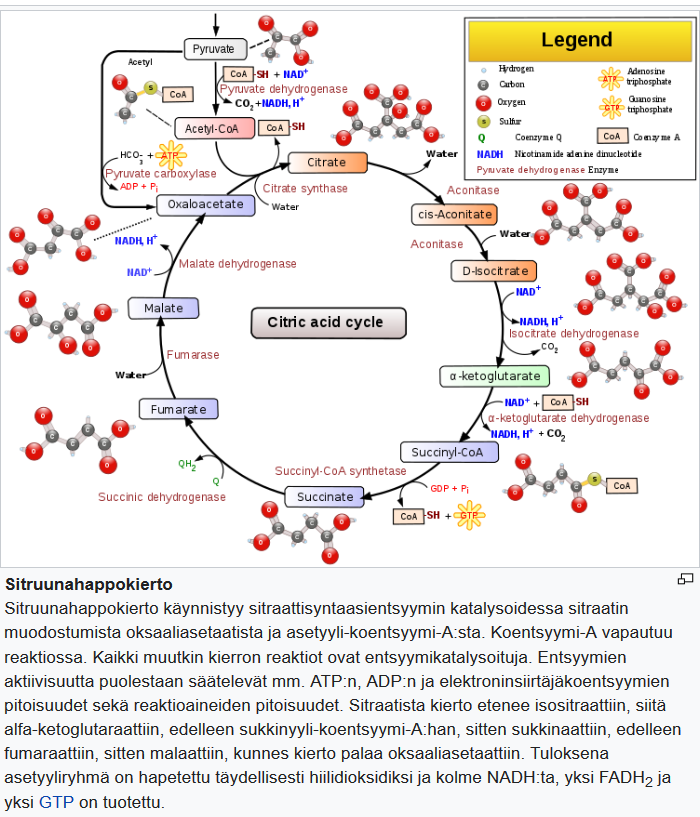
Espanjantauti, eli suuri influenssapandemia 1918-1920
Saatteeksi
Espanjaninfluenssa, joka tunnetaan myös nimillä espanjantauti, suuri influenssaepidemia tai vuoden 1918 influenssapandemia, oli poikkeuksellisen tappava maailmanlaajuinen pandemia, jonka aiheutti H1N1-influenssan A-alatyyppi.
Varhaisin dokumentoitu tapaus havaittiin maaliskuussa 1918 Kansasissa, Yhdysvalloissa. Tartuntoja rekisteröitiin huhtikuussa Ranskassa, Saksassa ja Isossa-Britanniassa. Kaksi vuotta myöhemmin lähes kolmasosa maailman väestöstä eli arviolta 500 miljoonaa ihmistä oli saanut tartunnan neljässä peräkkäisessä aallossa.
Maltilliset arviot espanjantautiin kuolleista vaihtelevat 17 miljoonasta 50 miljoonaan, mutta joidenkin arvioiden mukaan espanjantauti aiheutti jopa 100 miljoonan ihmisen kuoleman, mikä tekee siitä yhden kirjoitetun historian tappavimmista pandemioista. Vertailun vuoksi: SARS-CoV-2-epidemia on aiheuttanut yli 272 miljoonaa vahvistettua tartuntaa ja 5,3 miljoonaa kuolemaa. The Economist arvioi hiljattain, että todelliset luvut ovat moninkertaiset ja koronaan kuolleita voi olla jo yli 20 miljoonaa.
Nimi ”espanjalainen flunssa” on harhaanjohtava [6]. Pandemia puhkesi hieman ennen ensimmäisen maailmansodan loppua ilmeisesti Yhdysvalloissa. Sodanajan sensuurit tukahduttivat huonot uutiset sotaa käyvissä maissa moraalin ylläpitämiseksi, mutta sanomalehdet raportoivat vapaasti uudesta epidemiasta neutraalissa Espanjassa. Nämä uutiset loivat väärän kuvan Espanjasta uuden epidemian episentrumina, joten Espanjan ulkopuolelinen lehdistö otti käyttöön nimen ”espanjalainen flunssa”.
Rajalliset historialliset epidemiologiset tiedot tekevät pandemian maantieteellisestä alkuperästä epäselvän, ja alkuperäisestä leviämisestä on kilpailevia hypoteeseja [2].
Suurin osa influenssaepidemioista tappaa erityisesti vanhoja ja sairaita, mutta tämä pandemia aiheutti epätavallisen korkean nuorten aikuisten kuolleisuuden [7].
Tutkijoilla on joitain selitysmalleja tämän pandemian vakavuudelle ja nuorten korkealle kuolleisuudelle:
-
6 vuoden ilmastopoikkeama vaikutti taudinaiheuttajien kulkeutumiseen ja lisääntyneeseen leviämiseen vesistöjen kautta [8].
-
Virus oli nuorille tappavavampi, koska se laukaisi sytokiinimyrskyn, joka tuhosi nuorten aikuisten vahvemman immuunijärjestelmän [9]. Yleisesti infektio ei ilmeisesti ollut aggressiivisempi kuin aiemmat influenssakannat [10][11].
-
Aliravitsemus, ahtaat lääkärileirit ja sairaalat sekä huono hygienia, joita sota pahensi, lisäsivät bakteeriperäistä superinfektiota, mikä tappoi suurimman osan uhreista tyypillisesti pitkittyneen sairauden seurauksena [12][13].
-
Vuoden 1918 espanjantauti oli ensimmäinen kolmesta H1N1-influenssa A -viruksen aiheuttamasta influenssapandemiasta; viimeisin oli vuoden 2009 sikainfluenssapandemia [14][15].
-
Vuoden 1977 venäläisen flunssan aiheutti myös H1N1-virus, mutta se vaikutti enimmäkseen nuorempiin väestöihin [14][16].
- Käynnissä oleva COVID-19-pandemia, joka alkoi joulukuussa 2019 ja jonka aiheuttaa SARS-CoV-2, on vakavin pandemia sitten espanjantaudin [17].
Influenssan etymologia
 Kuva: Etusivu El Sol (Madrid), 28 May 1918: ”Kolmen päivän kuume on aiheuttanut 80 000 tartuntaa Madridissa. Kuningas on sairastunut”
Kuva: Etusivu El Sol (Madrid), 28 May 1918: ”Kolmen päivän kuume on aiheuttanut 80 000 tartuntaa Madridissa. Kuningas on sairastunut”
Espanjantautipandemia tunnettiin monilla eri nimillä. Osa nimistä oli vanhoja, osa uusia. Nimiin vaikutti paikka, aika ja konteksti. Vaihtoehtoisten nimien etymologia historiallistaa vitsauksen ja sen vaikutukset ihmisiin, jotka vasta vuosia myöhemmin oppivat, että näkymättömät virukset aiheuttivat influenssaa [18].
Sana influenssa on peräisin italian sanasta influenza joka on johdettu keskiaikaisen latinan sanasta influentia, Sana tarkoittaa ”vierailua” tai ”vaikutusta”. Käsitteet, kuten influenza di freddo, joka tarkoittaa ”kylmän vaikutusta”, ja influenza di stelle, joka tarkoittaa ”tähtien vaikutusta”, olivat todistettavasti käytössä jo 1300-luvulla. Jälkimmäinen viittaa sairauden syyhyn, jonka uskottiin johtuvan epäsuotuisista astrologisista olosuhteista.
Vuodesta 1504 alkaen influenssaksi on kutsuttu minkä tahansa suureen ihmisjoukkoon vaikuttavan sairauden ”vierailua” tai ”epidemiaa”. Vuonna 1743 Italiasta alkaneen ja kaikkialle Eurooppaan levinneen influenssaepidemian aikana sana saavutti englannin kielen, ja sen ääntäminen englantilaistui. 1800-luvun puolivälistä lähtien influenssaa on käytetty viittaamaan myös vakavasta vilustumisesta. Sanan lyhennetty muoto, ”flunssa”, tuli käyttöön ensimmäisen kerran vuonna 1839.
Muita influenssan nimityksiä ovat olleet epidemiakatarri, ranskan kielen la grippe, hikoilutauti ja, varsinkin kun viitataan vuoden 1918 pandemiakantaan, espanjakuume.
Dmitri Ivanovskyn ja sittemmin Ivanovskyn kokeet toistaneen Martinus Baijerinckin tutkimukset tupakkakasvin mosaiikkitaudista johtivat virusten nimeämiseen taudinaiheuttajina 1898, mutta vielä espanjantaudin aikana virusten luonne oli hyvin epäselvä. Influenssan syytä ei tunnettu.
Tieteellisen tiedon puute sai Sierra Leone Weekly Newsin (Freetown) ehdottamaan raamatullista selitystä heinäkuussa 1918 käyttäen kyselyä Exodus 16:sta muinaisen heprean kielellä: ”Yksi asia on varma – lääkärit ovat tällä hetkellä hämmästyneitä. Ehdotamme, että sen sijaan, että he kutsuisivat tautia influenssaksi, heidän pitäisi toistaiseksi kysyä Man hu: ”Mikä se on?” [20][21][22]
Violetti kuolema – sairautta kuvailevat nimet
Influenssan kaltaisten sairauksien puhkeamista dokumentoitiin vuosina 1916–1917 brittiläisissä sotisairaaloissa Étaplesissa Ranskassa [23] ja Englannin kanaalin toisella puolella Aldershotissa Englannissa.
Vuoden 1918 pandemian kanssa yhteisiä kliinisiä indikaatioita olivat oireiden nopea eteneminen kasvojen heliotrooppiseksi syanoosiksi. Tämä tyypillinen sinivioletti syanoosi vanhenevilla potilailla johti nimeen ”violetti kuolema” [24][25][26]. Aldershotin lääkärit kirjoittivat myöhemmin The Lancetissa: ”pneumokokin aiheuttama märkivä keuhkoputkentulehdus, jonka me ja muut kuvailimme vuosina 1916 ja 1917, on pohjimmiltaan vastaava kuin nykyisen pandemian aiheuttama influenssa” [27]. ”Märkivä keuhkoputkentulehdus” ei vielä liittynyt A/H1N1-virukseen [28], mutta se saattoi olla A/H1N1- viruksen esiaste [27][29][30].
Vuonna 1918 ’influenssaepideeminen’ (italiaksi: influenza, influence) sairaus [31], joka tunnettiin tuolloin myös nimellä ’grippi’ (ranska: la grippe, grasp),[32] havaittiin Kansasissa Yhdysvalloissa myöhään keväällä. Varhaiset raportit Espanjasta havaituista tartunnoista rekisteröitiin 21. toukokuuta [33][34]. Molemmista paikoista saadut raportit kutsuivat tautia ”kolmen päivän kuumeeksi” (fiebre de los tres días) [35][36][37].
Vaihtoehtoiset nimet
Monet vaihtoehtoiset nimet ovat eksonyymejä siinä, että uudet tartuntataudit näyttävät vierailta [38][39][40].
Tämä kuvio havaittiin jo ennen vuosien 1889–1890 pandemiaa, joka tunnetaan myös nimellä ”venäläinen flunssa”. Venäläiset kutsuivat epidemia ”kiinalaiseksi katarriksi”, saksalaiset kutsuivat sitä ”venäläiseksi tuholaiseksi”, kun taas italialaiset nimittivät sitä ”saksalaiseksi taudiksi” [41][42]. Näitä epiteettejä käytettiin uudelleen vuoden 1918 pandemiassa uusien epiteettien kanssa [43]. Tuoreempi esimerkki tällaisesta käytännöstä on Donald Trump, joka puhuu koronaviruksesta ”Kiinan viruksena”.
Espanjalainen influenssa
Espanjan ulkopuolella tauti nimettiin Espanjan influenssaksi [44][45]. 2. kesäkuuta 1918 The Times of London -lehti julkaisi Madridin kirjeenvaihtajan artikkelin, jonka otsikkona oli ”Espanjalainen epidemia”. Lehdessä kuvailtiin yli 100 000 uhria tartuttanutta ”tuntematonta tautia ja sen selvästi kauhistuttaa luonnetta” viittaamatta suoraan ”espanjalaiseen influenssaan” [46].
Kolme viikkoa myöhemmin The Times raportoi, että ”Kaikki pitävät sitä ”espanjalaisena influenssana tänä päivänä ” [47]. Pian tämän jälkeen The Timesissa ilmestyi Formamint-tablettien mainos ”espanjalaisen influenssan” estämiseksi [48] [49].
Kun tieto saapui Moskovaan, Pravda ilmoitti: ”Ispánka (espanjalainen nainen) on kaupungissa”, mikä teki ”espanjalaisesta naisesta” taudin toisen yleisen nimen [50].
Epidemia ei alkanut Espanjasta [51], mutta taudista raportointiin vaikutti sotaa käyvien maiden sodanaikainen sensuuri. Espanja oli neutraali maa, jolla ei ollut sota-aikaista propagandakoneistoa moraalin tukemiseksi [52][53], joten espanjalaiset sanomalehdet raportoivat vapaasti epidemiavaikutuksista, kuten kuningas Alfonso XIII:n sairastumisesta, mikä vahvisti mielikuvaa Espanjasta epidemian keskuksena [54]. Sotasensuuri oli niin toimivaa, että Espanjan terveysviranomaiset eivät tienneet, että epidemia aiheutti sairastumisia myös sen naapurimaissa [55].
Lokakuussa 1918 Journal of the American Medical Associationille osoitetussa ”Madridin kirjeessä” espanjalainen virkamies protestoi: ”Olimme yllättyneitä kuullessamme, että epidemia koettelee muitakin ja että tautia kutsutaan ”espanjalaiseksi otteeksi (grip)”. . Ja miksi espanja? …tämä epidemia ei syntynyt Espanjassa. Tämä on kirjattava historialliseksi todisteeksi” [56]. Mutta ennen kirjeen julkaisua, The Serbian Newspaper (Korfu) kirjoitti: ”Monet maat ovat syyttäneet tämän vieraan alkuperästä toisiaan. Jossain vaiheessa he sopivat syyttävänsä epidemian alkuperästä ystävällistä ja neutraalia Espanjaa…” [57].

Kuva: ”Espanjalainen influenssa,” ”kolmen päivän kuume, Rupert Blue, kirurgi, 28 syyskuuta 1918
Muut eksonyymit
Ranskalainen lehdistö käytti espanjantaudista alun perin nimitystä ”amerikkalainen flunssa”, mutta otti käyttöön ”espanjalaisen flunssan” liittolaisen vihamielisyyden vuoksi [58]. Keväällä 1918 brittiläiset sotilaat kutsuivat tautia ”Flanderin flunssaksi”, kun taas saksalaiset sotilaat käyttivät nimeä ”Flandern-Fieber” (Flanderin kuume) kuuluisan Belgiassa sijaitsevan taistelukentän mukaan, jossa monet sotilaat sairastuivat [43][40] ][59][60].
Senegalissa taudin nimi oli ”brasilian flunssa” ja Brasiliassa ”saksalainen flunssa” [61]. Espanjassa tauti tunnettiin myös nimellä ”ranskalainen flunssa” (gripe francesa) [51][6] tai ”Napolin sotilas” (Soldado de Nápoles) zarzuelan suositun kappaleen mukaan [b][58].
Espanjalainen flunssa (gripe española) on nykyään yleinen nimi Espanjassa[63], mutta se on siellä edelleen kiistanalainen [64][65]. Puolassa se oli ”bolshevikkien tauti”,[61][66] kun taas bolshevikit kutsuivat sitä ”kirgiisitaudiksi”[60].
Jotkut afrikkalaiset kutsuivat influenssaa ”valkoisen miehen sairaudeksi”, mutta Etelä-Afrikassa valkoiset miehet käyttivät myös etnofaulismia ”kaffersiekte” (lit. Neekeritauti) [43][67]. Japani syytti sumopainijoita siitä, että he toivat taudin kotiin ottelusta Taiwanissa kutsumalla sitä ”sumoflunssaksi” (Sumo Kaze), koska kolme huippupainijaa kuoli siellä [68][69][70].
Maailman terveysjärjestön vuonna 2015 julkaistut parhaat käytännöt estävät sosiaalisen leimaamisen. Kulttuurisesti leimaavia nimiä ei enää yhdistetä uusiin sairauksiin. ”espanjalainen flunssa” luetellaan ”vältettävien esimerkkien” alle [71][39][72]. Hyvin tuore esimerkki käytännön muuttumisesta on Intiassa kehittynyt SARS-CoV-2-viruksen ”intialainen variantti”, joka tunnetaan deltavarianttina, ja Etelä-Afrikassa havaittu ”eteläafrikkalainen variantti”, josta käytetään nimeä omikron.
Monet kirjoittajat välttelevät kutsumasta vuosien 1918-1920 pandemiaa espanjan influenssaksi [58] ja käyttävät muunnelmia sanasta ’1918–19/20 flunssa/influenssapandemia’ [73][74][75].
Paikalliset nimet
Joihinkin alkuperäiskansojen kielten endonyymeihin ei sisältynyt syyllistämistä tai uhrien häpeämistä. Tälle pandemialle ominaisia esimerkkejä ovat:
- pohjois-ndebele: ”malibuzwe” (kysykää taudista),
- swahili: ”ugonjo huo kichwa na kukohoa na kiuno” (pään, yskän ja selkärangan sairaus)[76],
- yao: ” chipindupindu’ (tauti, joka johtuu voiton tavoittelusta sodan aikana),
- otjiherero: ’kaapitohanga’ (sairaus, joka kulkee kuin luoti)[77] ja
- persiaksi: ’nakhushi-yi bad’ (tuulen sairaus) [78] ][79].
Muut nimet
Tämä epidemia tunnettiin yleisesti myös ”suurena influenssaepidemiana” [80][81] ”suuren sodan” jälkeen (yleinen nimi ensimmäiselle maailmansodalle ennen toista maailmansotaa)[9].
Ranskalaiset sotilaslääkärit kutsuivat tautia alun perin ”taudiksi 11” (maladie onze) [40]. Saksalaiset lääkärit vähättelivät sen vakavuutta kutsumalla sitä ”pseudo-influenzaksi”, kun taas Afrikassa lääkärit yrittivät saada potilaat suhtautumaan tautiin vakavammin kutsumalla sitä ”influenza veraksi” (latinaksi: vera, totta) [ 82].
Lastenlaulu vuosien 1889–1890 flunssapandemiasta [83] lyhennettiin ja mukautettiin hyppynaruloruksi, joka oli suosittu vuonna 1918 [84][85]. Se on metafora ”influenssan” leviävyydestä, jossa nimi leikattiin afereesiin ”Enza” (sanasa influenza)[86][87][88]:
I had a little bird,
its name was Enza.
I opened the window,
and in-flu-enza.
Historia
Aikajana
Ensimmäinen aalto 1918
 Kuva: Seattlen poliisit käyttivät valkoisia kangasmaskeja Espanjan flunssapandemian aikana joulukuussa 1918
Kuva: Seattlen poliisit käyttivät valkoisia kangasmaskeja Espanjan flunssapandemian aikana joulukuussa 1918
Perinteisesti pandemian katsotaan alkaneen 4. maaliskuuta 1918, kun Albert Gitchellin, Camp Funstonin armeijan kokin (Kansas, USA), tapaus rekisteröitiin. Joitain tapauksia tosin raportoitiin ennen maliskuuta [89].
Tauti oli havaittu Haskellin piirikunnassa jo tammikuussa 1918, mikä sai paikallisen lääkärin Loring Minerin kirjoittamaan varoituksen Yhdysvaltain kansanterveyspalvelun akateemisen aikakauslehden Public Health Reports toimittajille [9].
Muutaman päivän sisällä Camp Funstonin ensimmäisestä tapauksesta 522 miestä sairastui [90].
11. maaliskuuta 1918 virus oli saavuttanut New Yorkin Queensin [91]. Ennaltaehkäisevien toimien laiminlyöntiä maalis-huhtikuussa kritisoitiin myöhemmin [92]. Kun Yhdysvallat oli liittynyt ensimmäiseen maailmansotaan, tauti levisi nopeasti Camp Funstonista, muihin Yhdysvaltain armeijan varuskuntiin ja Eurooppaan. Taudista tuli epidemia Keskilännen, Itärannikon ja Ranskan satamissa huhtikuuhun 1918 mennessä ja länsirintamalla kuun puoliväliin mennessä [89].
Tämän jälkeen tauti levisi nopeasti muualle Ranskaan, Isoon-Britanniaan, Italiaan ja Espanjaan ja saavutti toukokuussa Breslaun ja Odessaan[89]. Brest-Litovskin sopimuksen allekirjoittamisen (maaliskuussa 1918) jälkeen Saksa alkoi vapauttaa venäläisiä sotavankeja, jotka sitten kuljettivat taudin Venäjälle[93].
Influenssa-aalto saapui Pohjois-Afrikkaan, Intiaan ja Japaniin toukokuussa, ja pian sen jälkeen se oli todennäköisesti levinnyt ympäri maailmaa, sillä Kaakkois-Aasiassa oli havaittiin tartuntoja huhtikuussa [94].
Kesäkuussa ilmoitettiin epidemiasta Kiinassa [95]. Saavuttuaan Australiaan heinäkuussa aalto alkoi laantua [94]. Ensimmäinen influenssa-aalto kesti vuoden 1918 ensimmäiseltä neljännekseltä ja oli suhteellisen lievä [96]. Kuolleisuusluvut eivät olleet merkittävästi normaalia kausi-influenssaa korkeammat [2]. Yhdysvalloissa ilmoitettiin noin 75 000 influenssaan liittyvää kuolemaa vuoden 1918 ensimmäisen kuuden kuukauden aikana verrattuna noin 63 000 kuolemaan samana ajanjaksona vuonna 1915 [97]. Madridissa Espanjassa alle 1 000 ihmistä kuoli influenssaan touko-kesäkuun 1918 välisenä aikana [98].
Karanteeneista ei raportoitu vuoden 1918 ensimmäisellä neljänneksellä. Ensimmäinen aalto aiheutti kuitenkin merkittävän häiriön ensimmäisen maailmansodan sotilasoperaatioissa: kolme neljäsosaa ranskalaisista joukoista, puolet brittijoukoista ja yli 900 000 saksalaista sotilasta sairastui [99].
Tappava toinen aalto alkoi vuoden 1918 toisella puoliskolla
 Kuva: American Expeditionary Force. Influenssan uhrit Yhdysvaltain armeijan leirin sairaalassa nro. 45 Aix-les-Bainsissa, Ranskassa, vuonna 1918
Kuva: American Expeditionary Force. Influenssan uhrit Yhdysvaltain armeijan leirin sairaalassa nro. 45 Aix-les-Bainsissa, Ranskassa, vuonna 1918
Pandemian toinen aalto alkoi elokuun toisella puoliskolla 1918. Se levisi Sierra Leonessa Bostoniin ja Freetowniin Brestistä saapuneiden laivojen kuljettamien amerikkalaisten ja ranskalaisjoukkojen mukana [99].
Boston Navy Yardista ja Camp Devensistä tauti eteni sotilaiden mukana muihin varuskuntiin. Muut Yhdysvaltain armeijan kohteet altistuivat kun joukkoja kuljetettiin Eurooppaan [100]. Joukkojen mukana tauti levisi seuraavien kahden kuukauden aikana koko Pohjois-Amerikkaan ja edelleen Keski- ja Etelä-Amerikkaan saavuttaen myös Brasilian ja Karibian [101].
Heinäkuussa 1918 Ottomaanien valtakunnassa havaittiin ensimmäiset tartunnat [102]. Freetownista pandemia levisi edelleen Länsi-Afrikan läpi rannikkoa, jokia ja siirtomaa-rautateitä pitkin sekä rautateiden päistä syrjäisimpiin yhteisöihin. Etelä-Afrikka vastaanotti toisen aallon syyskuussa. Se saapui Ranskasta syntyperäisiä eteläafrikkalaisia työläisiä kuljettavien laivojen mukana [101]. Se levisi ympäri Etelä-Afrikkaa ja Sambezin ulkopuolelle saavuttaen Etiopian marraskuussa [103].
Syyskuun 15. päivänä New Yorkissa raportoitiin ensimmäinen influenssakuolema [104]. Philadelphia Liberty Loans Parade, jossa edistettiin valtion joukkovelkakirjalainoja sodan menojen kattamiseksi Pennsylvaniassa 28. syyskuuta 1918, johti 12 000 ihmisen kuolemaan sen jälkeen, kun tauti levisi paraatiin osallistuneiden ihmisten keskuudessa [105].
Euroopasta toinen aalto pyyhkäisi Venäjälle ja lounais-Venäjältä koilliseen diagonaalisessa rintamassa. Pohjois-Venäjän intervention* vaikutuksesta toinen aalto levisi Arkangeliin ja edelleen koko Aasiaan. Venäjän sisällissota ja Trans-Siberian rautatie veivät taudin Iraniin (missä se levisi pyhän kaupungin Mashhadin kautta) ja Intiaan syyskuussa sekä Kiinaan ja Japaniin lokakuussa.[106]
*(Pohjois-Venäjän interventio oli osa ympärysvaltojen hanketta, jossa osallistuttiin Neuvosto-Venäjän vastaiseen taisteluun Venäjän sisällissodassa Venäjän valkoisten tukena vuosina 1918–1920. Interventioon osallistuivat Iso-Britannia, Yhdysvallat, Ranska ja Kanada.)
Aselevon (11. marraskuuta 1918) juhliminen aiheutti epidemioita myös Limassa ja Nairobissa, mutta joulukuussa aalto oli pääosin ohi [107]. Vuoden 1918 pandemian toinen aalto oli paljon tappavampi kuin ensimmäinen aalto.
Ensimmäinen aalto oli muistuttanut tyypillisiä flunssaepidemioita; suurimmassa vaarassa olivat sairaat ja vanhukset, kun taas nuoremmat, terveemmät ihmiset toipuivat helposti.
Lokakuussa 1918 kuolleisuus oli suurinta koko pandemian aikana [108]. USA:ssa ilmoitettiin noin 292 000 kuolemantapausta syys-joulukuun 1918 välisenä aikana, kun vastaava luku vuonna 1915 oli noin 26 000 [97]. Alankomaat raportoi yli 40 000 kuolemantapauksesta influenssan ja hengityselinsairauksien vuoksi. Bombay raportoi noin 15 000 kuolemantapauksesta [109]. Vuoden 1918 influenssapandemia oli erityisen tappava Intiassa, jossa arviolta 12,5–20 miljoonaa kuoli pelkästään vuoden 1918 viimeisellä neljänneksellä [96].
Kolmas aalto 1919
Tammikuussa 1919 flunssan kolmas aalto iski Australiaan, jossa se tappoi merikaranteenin poistamisen jälkeen noin 12 000 ihmistä ja levisi sitten nopeasti Euroopan ja Yhdysvaltojen halki, missä se viipyi kesäkuuhun 1919 asti 110][111][107]. Kolmas aalto iski rajuimmin Espanjaan, Serbiaan, Meksikoon ja Isoon-Britanniaan, mikä johti satojen tuhansien sairastuneiden kuolemaan [112].
Taudin kolmas aalto oli vähemmän vakava kuin toinen aalto, mutta silti paljon tappavampi kuin pandemian ensimmäinen aalto.
Yhdysvalloissa yksittäisiä epidemioita esiintyi joissakin kaupungeissa, kuten Los Angelesissa [113], New Yorkissa [1], Memphisissä, Nashvillessä, San Franciscossa ja St. Louisissa [114]. Kaiken kaikkiaan amerikkalaisten kuolleisuusluvut olivat kymmeniä tuhansia vuoden 1919 kuuden ensimmäisen kuukauden aikana [115].
Neljäs aalto 1920
Keväällä 1920 influenssapandemian neljäs aalto vyöryi New Yorkiin [1], Sveitsiin, Skandinaviaan [116] ja eräille Etelä-Amerikan saarille [117]. New York City raportoi 6 374 kuolemantapauksesta joulukuun 1919 ja huhtikuun 1920 välisenä aikana, mikä on lähes kaksi kertaa enemmän kuin kevään 1918 ensimmäisen aallon aikana [1].
Monissa USA:n kaupungeissa tauti oli raju. Detroitissa, Milwaukeessa, Kansas Cityssä, Minneapolisissa ja St. Louisissa kuolleisuus taudin neljänteen aaltoon oli korkeampi kuin vuonna 1918 [118].
Peru koki myöhäisen aallon vuoden 1920 alussa. Japanissa neljäs aalto kesti vuoden 1919 lopusta vuoden 1920 maaliskuulle [119]. Euroopassa saavutettiin myöhäinen huippu nfluenssapandemian neljännen aallon lyödessä viiteen maahan (Espanjaan, Tanskaan, Suomeen, Saksaan ja Sveitsiin) tammi-huhtikuussa 1920 [116].Espanjantauti Suomessa
Espanjantautia esiintyi Suomessa neljässä aallossa muutamien kuukausien välein. Se saapui maahan kesäkuussa 1918 laivojen mukana. Suomi oli saman vuoden alussa käydyn sisällissodan takia sekavassa tilassa, ja tauti levisi nopeasti. Kuolleisuus oli tässä vaiheessa vähäistä. Kesän päättyessä tautikin oli poissa.
Syksyllä 1918 espanjantauti palasi Suomeen entistä rajumpana tappaen runsaasti aikuisväestöä. Mitään tehokasta lääkitystä ei tuohon aikaan ollut, vaan helpotukseksi tarjottiin esimerkiksi kamferintippoja tai konjakkia. Lääkintähuolto oli muutenkin puutteellista ja lääkäreistä oli pulaa. Kansalaisia neuvottiin välttämään yleisötilaisuuksia ja pesemään käsiään huolellisesti välttääkseen tartunnan. Vuodenvaihteen aikoihin epidemia hiipui jälleen.
Keväällä 1919 Suomeen iski espanjantaudin kolmas aalto, joka oli ankarin muun muassa Helsingissä. Hautoja ei ehditty kaivaa riittävän nopeasti, kun kuolleisuus lisääntyi äkillisesti. Neljäs aalto tammi-helmikuussa 1920 vaikutti voimakkaimmin Lapissa. Inarissa joka kymmenes asukas menehtyi tuolloin espanjantautiin.
Lääkintöhallituksen saamien ilmoitusten perusteella on laskettu, että tautiin sairastui Suomessa yli 90 000 henkilöä vuonna 1918, yli 50 000 vuonna 1919 ja vielä yli 70 000 vuonna 1920 eli yhteensä ainakin 210 000 ihmistä. Espanjantautiin kuoli vuosina 1918–1920 arviolta noin 20 000 suomalaista.

Kuva: Amerikan Punaisen Ristin sairaanhoitajat hoitavat flunssapotilaita väliaikaisilla osastoilla, jotka on perustettu Oakland Municipal Auditoriumiin, 1918.
Pandemian loppu
Vuoteen 1920 mennessä pandemian aiheuttaneesta viruksesta tuli vähemmän tappava ja se aiheutti vain tavallista kausi-influenssaa.[120]
Pandemian mahdollinen alkuperä
Nimestään huolimatta historialliset ja epidemiologiset tiedot eivät pysty varmistamaan vuosien 1918-1920 espanjantautipandemian maantieteellistä alkuperää [2]. Pandemian synnystä on esitetty useita hyvin perusteltuja hypoteeseja. Varmuutta viruksen alkuperästä ei kuitenkaan ole.
Yhdysvallat
Ensimmäiset vahvistetut tautitapaukset olivat peräisin Yhdysvalloista. Historioitsija Alfred W. Crosby totesi vuonna 2003, että influenssa sai alkunsa Kansasista [121]. Kirjailija John M. Barry kuvaili taudin puhkeamista tammikuussa 1918 Haskell Countyssa, Kansasissa, vuoden 2004 artikkelissaan [9].
Evoluutiobiologian professori Michael Worobeyn johtamassa vuonna 2018 tehdyssä kudoslevyjä ja lääketieteellisiä raportteja koskevassa tutkimuksessa löydettiin todisteita Kansasista peräisin olevaa tautia vastaan, koska tapaukset olivat lievempiä ja niissä kuoli vähemmän ihmisiä kuin New Yorkissa saman ajanjakson infektioissa. Tutkimuksessa löydettiin fylogeneettisten analyysien avulla todisteita siitä, että viruksella oli todennäköisesti pohjoisamerikkalainen alkuperä, vaikka tutkimus ei päätynyt konklusiiviseen ratkaisuun.
Viruksen hemagglutiniiniglykoproteiinit viittaavat siihen, että se oli kehittynyt kauan ennen vuotta 1918. Muut tutkimukset viittaavat siihen, että H1N1-viruksen muunnos kehittyi todennäköisesti vuonna 1915 tai sen tienoilla [122].

Kuva: Edvard Munch (1863-1944) – Omakuva espanjantaudissa (1919)
Eurooppa

Kuva: Egon Schiele (1880-1918) – Die Familie (The Family), jonka hän maalasi vaimon kuoleman jälkeen vain päiviä ennen kuin hän itse kuoli [123]
Oxfordin mukaan samanlainen influenssaepidemia puhkesi maaliskuussa 1917 armeijan kasarmeissa Aldershotissa [126]. Armeijan patologit tunnistivat myöhemmin nämä varhaiset tautiaallot samaksi taudiksi kuin espanjantauti [127][124]. Etaplesin ruuhkainen leiri ja sairaala olivat ihanteellisia ympäristöjä hengitystieviruksen leviämiselle. 100 000 sotilasta kulki leirin läpi päivittäin; sairaalassa hoidettiin tuhansia myrkkykaasuiskujen uhreja ja muita haavoittuneita.
Etaplesissa oli myös sikala, ja siipikarjaa tuotiin säännöllisesti ympäröivistä kylistä leirin sotilaiden ravinnoksi. Oxford ja hänen tiiminsä uskovat, että linnuissa oleva esiastevirus mutatoitui ja siirtyi sitten rintaman lähellä pidettyihin sikoihin [126][127].
Vuonna 2016 Journal of the Chinese Medical Association -lehdessä julkaistussa raportissa löydettiin todisteita siitä, että vuoden 1918 virus oli kiertänyt Euroopan armeijoissa kuukausia ja mahdollisesti vuosia ennen vuoden 1918 pandemiaa [128].
Politologi (politiikan tutkija) Andrew Price-Smith julkaisi Itävallan arkistojen tietoja, joiden mukaan influenssa alkoi Itävallassa vuoden 1917 alussa [129]. Vuonna 2009 tehdyssä influenssaa ja muita hengityselinten viruksia käsittelevässä tutkimuksessa havaittiin, että espanjantautikuolleisuus saavutti huippunsa samanaikaisesti kahden kuukauden aikana loka-marraskuussa 1918 kaikissa neljässätoista analysoidussa Euroopan maassa, mikä on ristiriidassa sen mallin kanssa, ettå virus olisi kehittynyt Euroopassa ja levinnyt Euroopasta muualle maailmaan [130].
Kiina
Vuonna 1993 Pasteur-instituutin johtava espanjantautiasiantuntija Claude Hannoun väitti, että espanjantaudin esiastevirus oli todennäköisesti peräisin Kiinasta. Se mutatoitui Yhdysvalloissa lähellä Bostonia ja levisi sieltä Ranskaan ja Euroopan taistelukentille, sekä edelleen liittoutuneiden sotilaiden ja merimiesten levittämänä muualle Eurooppaan ja maailmaan [131]. Hanoun piti useita vaihtoehtoisia alkuperähypoteesejä, kuten Espanja, Kansas ja Brest, mahdollisina, mutta epätodennäköisinä [131].
Vuonna 2014 historioitsija Mark Humphries arveli, että pandemian syynä saattoi olla 96 000 kiinalaisen työläisen mobilisointi brittiläisten ja ranskalaisten linjojen takana.
Humphries (Memorial University of Newfoundland St. John’s) perusti johtopäätöksensä uusiin asiakirjoihin. Hän löysi arkistoista todisteita siitä, että marraskuussa 1917 Pohjois-Kiinassa (josta työläiset tulivat) levisi hengityselinsairaus, jonka Kiinan terveysviranomaiset tunnistivat vuotta myöhemmin identtiseksi espanjantaudin kanssa [132][133]. Kudosnäytteitä ei kuitenkaan ole säilynyt vertailevan tutkimuksen tekemiseksi [134]. Kiinalaisten työläisten kulkeman reitin varrelta on säilynyt joitain raportteja espanjantautia muistuttavista tartunnoista. Reitti kulki Eurooppaan Pohjois-Amerikan kautta [134].
Kiina oli yksi harvoista maailman alueista, jossa espanjantautiepidemia aiheutti vain lievän flunssa-aallon. Tutkimukset ovat dokumentoineet verrattain lievän flunssakauden vuonna 1918 [135][136][137]. Vaikka tiedot ovat puutteellisia, tämä on johtanut spekulaatioon, että espanjantauti kehittyi Kiinassa [137][138]. Kiinan pientä influenssakuolleisuutta selittäisi väestön immuniteetti ko. Virukselle [139][137].
Journal of the Chinese Medical Association -lehdessä vuonna 2016 julkaistu raportti ei löytänyt todisteita siitä, että vuoden 1918 virus olisi saapunut Eurooppaan kiinalaisten tai kaakkois-aasialaisten sotilaiden ja työntekijöiden mukana. Sen sijaan löytyi todisteita espanjantautiviruksen leviämisestä Euroopassa ennen pandemian puhkeamista [128].
Vuoden 2016 tutkimuksessa pääteltiin, että kiinalaisten ja kaakkois-aasialaisten työntekijöiden alhainen influenssakuolleisuus Euroopassa (arviolta yksi tuhannesta) tarkoitti sitä, että vuoden 1918 tappava influenssapandemia ei voinut olla peräisin näiltä työntekijöiltä [128].
Lisätodisteena kiinalaisten työntekijöiden levittämää tautia vastaan oli se, että työntekijät saapuivat Eurooppaan monia reittejä pitkin, joiden varrella ei havaittu tartuntoja ja taudin leviämistä, minkä vuoksi kiinalaiset työläiset eivät todennäköisesti olleet alkuperäisiä isäntiä [122].
Influenssaepidemian epidemiologia ja patologia
Tartunnat ja mutaatiot
 Kuva: Kun Yhdysvaltain joukot lähtivät suurella joukolla sotimaan Eurooppaan, he kantoivat mukanaan espanjantautia
Kuva: Kun Yhdysvaltain joukot lähtivät suurella joukolla sotimaan Eurooppaan, he kantoivat mukanaan espanjantautia
Viruksen tarttuvuusluku (R0) oli 2–3 [140]. Ensimmäisen maailmansodan aikaiset massiiviset joukkojen liikkeet ja tiiviisti asuneet ihmiset jouduttivat pandemiaa lisäten tartuntoja ja virukseen syntyneitä mutaatioita.
Sota saattoi heikentää ihmisten vastustuskykyä virukselle. Jotkut arvelevat, että aliravitsemus ja taistelujen aiheuttama stressi heikensivät sotilaiden immuunijärjestelmää, mikä lisäsi heidän infektioherkkyyttään [141][142]. Merkillepantava tekijä espanjantaudin maailmanlaajuisessa leviämisessä oli matkustamisen tehostuminen ja lisääntynyt matkustaminen. Modernit kuljetusjärjestelmät (junat, valtamerilaivat) helpottivat sotilaiden, merimiesten ja siviilimatkailijoiden liikkumista ja tehostivat taudin leviämistä.[143]
Espanjantaudin leviämistä tehosti myös sotasensuuri, epidemian julkinen vähättely ja hallitusten valheet, mikä jätti väestön huonosti valmistautuneeksi epidemiaan [144]. Espanjantaudin toisen aallon vakavuuden on katsottu johtuvan ensimmäisen maailmansodan olosuhteista [145].
Siviilielämässä luonnonvalinta suosii lievän sairauden aiheuttavaa viruskantaa: hyvin sairaat jäävät kotiin, ja lievästi sairaat jatkavat elämäänsä levittäen lievempää viruskantaa. Juoksuhaudoissa luonnonvalinta kääntyi päinvastaiseksi. Sotilaat, joilla oli lievä tauti, pysyivät paikoillaan, kun taas vakavasti sairaat lähetettiin täyteen ahdetuissa junissa täpötäysiin kenttäsairaaloihin levittämään tappavampaa viruskantaa. Toisen aallon alettua espanjantauti levisi nopeasti uudelleen ympäri maailman. Espanjantaudista saatujen havaintojen seurauksena terveysviranomaiset etsivät nykyään pandemioissa tappavampia viruskantoja alueilta, joissa tapahtuu sosiaalisia mullistuksia [146]. Sodat ja köyhyys luovat puitteet, joissa virus leviää tehokkaasti ja siitä kehittyy vakavampia virusmuunnoksia.
Se, että useimmat ensimmäisen aallon infektioista toipuneet olivat tulleet immuuniksi, osoitti, että toisen aallon aiheutti sama virus. Tämä näkyi dramaattisimmin Kööpenhaminassa, jossa yhteenlaskettu kuolleisuusaste oli vain 0,29 % (0,02 % ensimmäisessä aallossa ja 0,27 % toisessa aallossa) johtuen altistumisesta vähemmän tappavalle ensimmäiselle aallolle [147].
Ensimmäisen aallon välttäneelle väestölle toinen aalto oli paljon tappavampi kuin ensimmäinen aalto. Erityisen haavoittuvia olivat nuoret hyväkuntoiset aikuiset, kuten sotilaat juoksuhaudoissa [148]. Kun tappava toinen aalto iski vuoden 1918 lopulla, uudet tapaukset vähenivät äkillisesti. Esimerkiksi Philadelphiassa 4 597 ihmistä kuoli 16. lokakuuta päättyneellä viikolla, mutta marraskuun 11. päivään mennessä influenssa oli melkein kadonnut kaupungista. Yksi selitys taudin kuolleisuuden nopealle vähenemiselle on se, että lääkärit tehostivat keuhkokuumeen ehkäisyä ja hoitoa. John Barry kuitenkin totesi vuoden 2004 kirjassaan The Great Influenza: The Epic Story of the Deadliest Plague In History, että tutkijat eivät ole löytäneet todisteita tämän näkemyksen tueksi [9].
Toisen teorian mukaan vuoden 1918 virus mutatoitui erittäin nopeasti vähemmän tappavaksi kannaksi. Tällainen influenssan kehitys on yleistä: patogeenisillä viruksilla on taipumus tulla vähemmän tappaviksi ajan myötä, koska vaarallisempien kantojen isännät kuolevat[ 9]. Jotkut kuolemaan johtaneet tapaukset jatkuivat maaliskuuhun 1919 ja tappoivat yhden pelaajan Stanley Cupin finaalissa 1919.
Merkit ja oireet
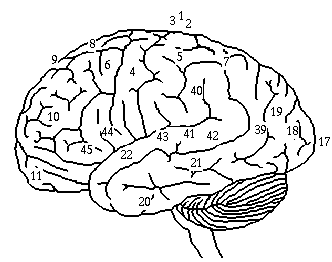
 Kuva: Flunssaoireet, Yhdysvaltojen armeija
Kuva: Flunssaoireet, Yhdysvaltojen armeija
Suurin osa tartunnan saaneista koki vain tyypillisiä flunssaoireita, kuten kurkkukipua, päänsärkyä ja kuumetta, erityisesti espanjantaudin ensimmäisen aallon aikana [149]. Toisen aallon aikana tauti oli kuitenkin paljon vakavampi. Bakteeriperäinen keuhkokuume oli usein kuoleman syy [149].
Tämä vakavampi virustyyppi aiheutti heliotrooppisen syanoosin, jossa poskipäihin muodostui ensin kaksi ”mahonkitäplää”. Ihomuutokset levisivät muutaman tunnin kuluessa värjäten kasvot sinertäviksi, mitä seurasi ihon mustuminen ensin raajoissa ja sitten mustumisen leviäminen edelleen vartaloon [149]. Tämän jälkeen kuolema seuraisi tunneissa tai päivissä, koska keuhkot täyttyivät nesteellä [149].
Muita taudin raportoituja merkkejä ja oireita olivat:
- spontaani suun ja nenäverenvuoto,
- odottavien naisten keskenmeno,
- erikoiset hajuaistimukset,
- hampaiden ja hiusten lähtö,
- delirium,
- huimaus,
- unettomuus,
- kuulon tai hajuaistin menetys,
- näön hämärtyminen ja värinäön heikkeneminen [149].
Eräs tarkkailija kirjoitti: ”Yksi silmiinpistävimmistä komplikaatioista oli limakalvojen verenvuoto, erityisesti nenästä, vatsasta ja suolesta. Verenvuotoa korvista ja peteekaalisia (petekia tarkoittaa verenpurkaumaa) verenvuotoja esiintyi myös iholla” [150]. Oireiden vakavuuden uskottiin johtuvan sytokiinimyrskystä [96]. Suurin osa kuolemista johtui bakteeriperäisestä keuhkokuumeesta [151][152][153], joka on yleinen sekundaarinen influenssaan liittyvä infektio. Tämä keuhkokuume johtui leisistä ylempien hengitysteiden bakteereista, jotka pääsivät keuhkoihin uhrien vaurioituneiden keuhkoputkien kautta [154].
Virus myös tappoi ihmisiä suoraan aiheuttamalla verenvuotoa ja turvotusta keuhkoissa [153]. Nykyaikainen analyysi on osoittanut viruksen olevan erityisen tappava, koska se voi laukaista sytokiinimyrskyn (immuunijärjestelmän ylireagoinnin) [9].
Eräsa tutkijaryhmä sai viruksen talteen jäädytettyjen uhrien ruumiista ja transfektoi eläimiä sillä. Eläimet kärsivät nopeasti etenevästä hengitysvajauksesta ja kuolivat sytokiinimyrskyn seurauksena. Nuorten aikuisten voimakkaiden immuunireaktioiden oletettiin tuhonneen kehoa, kun taas lasten ja keski-ikäisten aikuisten heikommat immuunireaktiot aiheuttivat vähemmän kuolemia näiden ryhmien keskuudessa [155].
Väärä diagnoosi
Koska taudin aiheuttanut virus oli liian pieni mikroskoopilla nähtäväksi, oikean diagnoosin tekemisessä oli ongelmia [156]. Haemophilus influenzae -bakteeria luultiin virheellisesti taudin syyksi, koska se oli tarpeeksi suuri mikroskoopilla nähtäväksi ja sitä esiintyi monilla, joskaan ei kaikilla potilailla [156]. Tästä syystä tätä basillia vastaan käytetty rokote ei tehnyt infektiosta harvinaisempaa, mutta vähensi infektioon kuolleisuutta [157].
Kuolettavan toisen aallon aikana pelättiin myös, että kyseessä oli rutto, denguekuume tai kolera [158]. Eräs yleinen virhediagnoosi oli lavantauti, joka levisi usein yhteiskunnallisen kaaoksen ja epähygieenisten olojen seurauksena, kuten Venäjällä lokakuun vallankumouksen jälkimainingeissa [158].
Chilessä maan eliitin näkemys oli, että kansakunta oli vakavassa taantumassa, ja siksi lääkärit olettivat, että tauti oli huonon hygienian aiheuttama lavantauti, eikä tarttuva tauti. Tämä näkemys johti taudin tehokkaampaan leviämiseen, koska tarttuvaa tautia ei yritetty estää leviämästä joukkokokoontumisia rajoittamalla [158].
 Kuva: Yskä ja aivastelu levittävät sairauksia. Yhdysvaltain kansanterveysmainos espanjatautiepidemian vaaroista ensimmäisen maailmansodan aikana.
Kuva: Yskä ja aivastelu levittävät sairauksia. Yhdysvaltain kansanterveysmainos espanjatautiepidemian vaaroista ensimmäisen maailmansodan aikana.
Ilmaston rooli
Tutkimukset ovat vahvistaneet, että espanjantaudin uhrien immuunijärjestelmää heikensivät epäsuotuisat ilmasto-olosuhteet, jotka olivat erityisen kylmiä ja märkiä pitkiä aikoja pandemian aikana. Tämä vaikutti erityisesti ensimmäisen maailmansodan sotilaisiin, jotka olivat alttiina jatkuville sateille ja keskimääräistä alhaisemmille lämpötiloille konfliktin ja erityisesti pandemian toisen aallon aikana.
Korkearesoluutioiset Harvardin yliopistossa ja Mainen yliopiston ilmastonmuutosinstituutissa analysoidut ilmastotiedot yhdistettynä erittäin yksityiskohtaisiin kuolleisuustietoihin tunnistivat vakavan ilmastopoikkeaman, joka vaikutti Eurooppaan vuosina 1914–1919. Useat ympäristöindikaattorit vaikuttivat suoraan epidemian vakavuuteen ja leviämiseen [8].
Sademäärän merkittävä lisääntyminen vaikutti koko Eurooppaan pandemian toisen aallon aikana syyskuusta joulukuuhun 1918.
Kuolleisuusluvut seuraavat tarkasti sateiden lisääntymistä ja lämpötilan laskua. Tälle on ehdotettu useita selityksiä, kuten se, että alhaisemmat lämpötilat ja lisääntynyt sademäärä loivat ihanteelliset olosuhteet viruksen replikaatiolle ja leviämiselle samalla kun ne vaikuttavat negatiivisesti sotilaiden ja muiden kolealle säälle altistuneiden ihmisten immuunijärjestelmään; tämä lisäsi todennäköisyyttä sekä virusten että pneumokokkien aiheuttamille samanaikaisille infektioille, joiden on dokumentoitu vaikuttaneen suureen osaan pandemian uhreista (viidennes sairastuneista, joiden kuolleisuusaste oli 36 prosenttia) [159][160][161][162][163].
Kuusi vuotta kestänyt ilmastopoikkeama (1914–1919) toi kylmää, merellistä ilmaa Eurooppaan muuttaen sen säätä dramaattisesti, kuten silminnäkijöiden kertomukset ja instrumentaaliset asiakirjat osoittavat. Ilmastopoikkeama jatkui Gallipolin kampanjaan asti Turkissa, jossa ANZAC-joukot kärsivät erittäin kylmästä ilmastosta. Ilmastopoikkeama vaikutti todennäköisesti vesistöjä ulosteillaan saastuttavien H1N1-lintujen migraatioon. Tartunnat saavuttivat syksyllä 60 % tason [164][165] [166]. Ilmastopoikkeama on liitetty ihmisen aiheuttamaan ilmakehän pölyn lisääntymiseen jatkuvien pommitusten vuoksi; pölyhiukkasten (pilvien kondensaatioytimien) aiheuttama lisääntynyt nukleaatio lisäsi sademäärää [167][168][169].
Vastatoimet
Julkinen terveydenhuolto
 Kuva: Syyskuussa 1918 Punainen Risti suositteli kaksikerroksisia sideharsonaamioita ”ruton” leviämisen estämiseksi [170].
Kuva: Syyskuussa 1918 Punainen Risti suositteli kaksikerroksisia sideharsonaamioita ”ruton” leviämisen estämiseksi [170].

Kuva: 1918 Chicagon sanomalehtien otsikot kertoivat influenssaepidemian lieventämisstrategioista, kuten ilmanvaihdon lisäämisestä, pidätyksistä kasvonaamion käyttämättä jättämisen vuoksi, rokotuksista, väkijoukon koon rajoituksista, yritysten valikoivasta sulkemisesta ja ulkonaliikkumiskielloista [171]. Kun lokakuun tiukat rajoitustoimenpiteet osoittivat jonkin verran menestystä, aselepopäivän juhliminen marraskuussa ja kiitospäivän vapautunut tunnelma johtivat taudin elpymisen [171].
Vaikka vuonna 1918 oli olemassa järjestelmiä, joilla varoitettiin kansanterveysviranomaisia tartuntojen leviämisestä, ne eivät yleensä huomioineet influenssaa, mikä johti viivästyneeseen reagointiin [172].
Influenssaepidemian vastaisiin toimenpiteisiin kuitenkin ryhdyttiin: merikaranteenit julistettiin monille saarille, kuten Islanti, Australia ja Amerikan Samoa [172]. Espanjantaudin vuoksi otettiin käyttöön sosiaalisia etäisyyksiä koskevia toimenpiteitä, kuten koulujen, teatterien ja jumalanpalveluspaikkojen sulkemiset, julkisen liikenteen rajoittaminen ja joukkokokoontumisten kieltäminen [173].
Kasvomaskien käyttäminen yleistyi joissakin paikoissa, kuten Japanissa, vaikka niiden tehokkuudesta käytiin kiivasta keskustelua [173]. Kasvosuojien käyttöä vastusti mm. San Franciscon Anti-Mask League.
Myös rokotteita kehitettiin, mutta koska ne perustuivat bakteereihin eivätkä espanjantautia aiheuttavaan virukseen, ne auttoivat vain sekundaarisissa infektioissa [173]. Rajoitusten tosiasiallinen täytäntöönpano vaihteli [174]. New Yorkin terveysasioista vastaava komissaari määräsi yrityksiä avaamaan ja sulkemaan palveluja porrastetusti metrojen ruuhkautumisen välttämiseksi [175].
Myöhemmin tehdyssä tutkimuksessa havaittiin, että epidemian vastaiset toimenpiteet, kuten joukkokokoontumisten kieltäminen ja kasvonaamioiden käyttö leikkasivat kuolleisuutta jopa 50 prosenttia, mutta hyöty saavutettiin vain, jos vastatoimet otettiin käyttöön epidemian varhaisessa vaiheessa eikä niitä poistettu ennenaikaisesti [176].
Lääkehoito
Koska viruksen hoitoon ei ollut antiviraalisia viruslääkkeitä tai antibiootteja sekundaaristen bakteeri-infektioiden hoitoon, lääkärit luottivat satunnaiseen valikoimaan lääkkeitä, joiden tehokkuus taudin hoidossa vaihteli. Espanjantautia hoidettiin aspiriinilla, kiniinillä, arseenilla, digitaliksella, strykniinillä, kamferitipoilla, epsom-suoloilla, risiiniöljyllä ja jodilla [177]. Hoidossa sovellettiin myös traditionaalisen lääketieteen menetelmiä, kuten verenlaskua, ayurvedaa ja kampoa.[178]
Tiedon saanti
Ensimmäisen maailmansodan vuoksi monet maat harjoittivat sodanaikaista sensuuria ja estivät espanjantautipandemiasta raportoinnin [179]. Esimerkiksi italialaista sanomalehteä Corriere della Sera’a kiellettiin raportoimasta päivittäisiä kuolonuhrien määriä [179]. Sen ajan sanomalehdet olivat myös yleisesti paternalistisia ja huolissaan joukkopanikista [179].
Väärä tieto levisi myös taudin mukana. Irlannissa uskottiin, että Flanders Fieldsin joukkohaudoista nousi haitallisia kaasuja ja ”tuulet puhaltavat niitä kaikkialle maailmaan” [180]. Huhuttiin, että saksalaiset olivat influenssan takana esimerkiksi myrkyttämällä Bayerin valmistaman aspiriinin tai vapauttamalla myrkkykaasua U-veneistä [181].
Kuolleisuus
Katso: List of Spanish flu cases
Koko maailma
 Kuva: Ero vuoden 1918 epidemian ja kausiepidemioiden influenssakuolleisuuden ikäjakaumien välillä – kuolemat 100 000 henkeä kohti kussakin ikäryhmässä, Yhdysvallat, pandemiavuosien 1911–1917 (katkoviiva) ja pandemiavuoden 1918 (yhtenäinen viiva) välillä [182]
Kuva: Ero vuoden 1918 epidemian ja kausiepidemioiden influenssakuolleisuuden ikäjakaumien välillä – kuolemat 100 000 henkeä kohti kussakin ikäryhmässä, Yhdysvallat, pandemiavuosien 1911–1917 (katkoviiva) ja pandemiavuoden 1918 (yhtenäinen viiva) välillä [182]

Kuva: Kolme pandemia-aaltoa: viikoittainen yhdistetty kuolleisuus influenssa- ja keuhkokuumeeseen, Yhdistynyt kuningaskunta, 1918–1919[183]
Espanjantauti levisi ~500 miljoonaan ihmiseen. Sairastuneiden määrä vastasi noin kolmannesta maailman väestöstä [2]. Arviot siitä, kuinka monta tartunnan saaneista ihmistä kuoli, vaihtelevat suuresti, mutta vuosien 1918-1920 suurta influenssapandemiaa (espanjantautia) pidetään yhtenä historian tappavimmista pandemioista [184][185].
-
Vuodelta 1927 peräisin olevan varhaisen arvion mukaan kuolleisuus maailmassa oli 21,6 miljoonaa [4].
-
Vuoden 1991 arvion mukaan virus tappoi 25–39 miljoonaa ihmistä [96].
-
Vuoden 2005 arvion mukaan kuolleiden määrä on 50 miljoonaa (noin 3 % maailman väestöstä) ja mahdollisesti jopa 100 miljoonaa (yli 5 %) [150][186].
-
American Journal of Epidemiology -lehdessä vuonna 2018 julkaistussa uudelleenarvioinnissa kuolleiden kokonaismääräksi arvioitiin noin 17 miljoonaa [4][187].
Kun maailman väkiluku oli espanjantaudin aikaan 1,8–1,9 miljardia [188], arviot vastaavat 1–6 prosenttia koko väestöstä. Vuonna 2009 tehdyssä influenssaa ja muita hengitystieviruksia koskevassa neljäntoista Euroopan maan tietoihin perustuvassa tutkimuksessa, arvioitiin yhteensä 2,64 miljoonan espanjantaudin aiheuttamaa kuolemaa Euroopassa pandemian suuren vaiheen 1918–1919 aikana. Vuosina 1991, 2002 ja 2006 Euroopassa kuolleiden määräksi laskettiin 2–2,3 miljoonaa. Tämä vastaa noin 1,1 prosentin kuolleisuutta Euroopan väestöstä (joka oli noin 250 miljoonaa vuonna 1918). Arvio on huomattavasti korkeampi kuin USA:n kuolleisuus, minkä kirjoittajat olettavat johtuvan sodan vakavista vaikutuksista Euroopassa 130]. Yhdistyneen kuningaskunnan ylikuolleisuuden on arvioitu olleen 0,28–0,4 prosenttia, mikä on selvästi alle eurooppalaisen keskiarvon [4].
Intiassa kuoli noin 12–17 miljoonaa ihmistä, eli ~5 % väestöstä [189]. Kuolleiden määrä Intian brittihallinnossa olevilla alueilla oli 13,88 miljoonaa [190]. Toisen arvion mukaan kuolleita oli Intiassa ainakin 12 miljoonaa [191]. Vuosien 1911 ja 1921välinen vuosikymmen oli ainoa väestönlaskentajakso, jolloin Intian väkiluku laski. Tämän arvellaan johtuvan pääasiassa espanjantaudin vaikutuksista [192][193]. Vaikka Intiaa kuvataan yleisesti espanjantaudista pahiten kärsineeksi maaksi, ainakin yhdessä tutkimuksessa väitetään, että vuonna 1918 havaitut erittäin korkeat ylikuolleisuusluvut voivat osittain selittyä muilla tekijöillä. Tämä tutkimus perustaa arvionsa vuoden 1917 epätavallisen korkeaan kuolleisuuteen ja laajaan alueelliseen vaihteluun (vaihteluväli 0,47 % – 6,66 % [4].
Vuonna 2006 The Lancet -lehdessä julkaistussa tutkimuksessa pääteltiin, että Intian provinssien ylikuolleisuusaste vaihteli 2,1 prosentista 7,8 prosenttiin, ja todettiin: ”Silloin kommentaattorit katsoivat tämän valtavan vaihtelun johtuvan ravitsemustilan eroista ja lämpötilan vuorokausivaihteluista” [194].
-
Suomessa espanjantautiin kuoli arviolta 20 000 noin 210 000 tartunnan saaneesta [195]
-
Ruotsissa kuoli 34 000 [196]
-
Japanissa sairastui 23 miljoonaan ihmistä ja ainakin 390 000 sairastuneista kuoli [197]
-
Hollannin Itä-Intiassa (Indonesia) 30 miljoonan väestöstä 1,5 miljoonan oletetaan kuolleen espanjantautiin [198]
-
Tahitissa 13 % väestöstä kuoli kuukauden aikana
-
Länsi-Samoalla 22 % 38 000 asukkaasta kuoli kahden kuukauden aikana [199]
-
Ottomaanien valtakunnan pääkaupungissa Istanbulissa espanjantautiin kuoli 6 403–10 000 [200][102] ihmistä, mikä antaa kaupungin kuolleisuusasteeksi vähintään 0,56 % [200]
- Uudessa-Seelannissa espanjantauti tappoi arviolta 6 400 pakehaa (”ensisijaisesti eurooppalaista syntyperää olevaa uusiseelantilaista”) ja 2 500 alkuperäiskansan maoria kuudessa viikossa. Maorien kuolleisuus oli kahdeksankertainen eurooppalaistaustaisiin verrattuna [201][202]
-
Yhdysvalloissa noin 28 % 105 miljoonan väestöstä sai tartunnan ja 500 000 – 850 000 kuoli (0,48 – 0,81 prosenttia väestöstä) [203][204][205]
-
Amerikan intiaaniheimot kärsivät erityisen kovasti. Four Cornersin alueella rekisteröitiin 3 293 kuolemaa intiaanien keskuudessa [206]
-
Alaskassa kuoli kokonaisia inuiittien kyläyhteisöjä [207]
-
Kanadassa kuoli 50 000 [208]
-
Brasiliassa kuoli 300 000, mukaan lukien presidentti Rodrigues Alves [209].
-
Britanniassa kuoli jopa 250 000
-
Ranskassa yli 400 000 ihmistä [210]
-
Ghanassa influenssaepidemia tappoi ainakin 100 000 ihmistä [211]
-
Tafari Makonnen (eli Haile Selassie, Etiopian keisari) oli yksi ensimmäisistä etiopialaisista, joka sairastui influenssaan, mutta selvisi hengissä [212][213] Monet hänen aiheistaan eivät selvinneet; arviot kuolemantapauksista pääkaupungissa Addis Abebassa vaihtelevat 5 000 – 10 000 välillä, mutta voivat olla korkeammatkin [214]
- Kuolonuhrien määräksi Venäjällä on arvioitu 450 000, vaikka epidemiologit, jotka ehdottivat tätä lukua, kutsuivat sitä ”laukaukseksi pimeässä”[96] Jos arvio pitää paikkansa, Venäjä menetti noin 0,4 % väestöstään, mikä tarkoittaa, että se kärsi Euroopan alhaisimmista influenssakuolleisuudesta. Toisessa tutkimuksessa tätä lukua pidetään epätodennäköisenä, koska maa oli sisällissodan kourissa ja jokapäiväisen elämän infrastruktuuri oli rikki; tutkimuksen mukaan Venäjän kuolonuhrien määrä oli lähempänä kahta prosenttia eli 2,7 miljoonaa ihmistä [215]
Pandemian vaurioittamat yhteisöt
 Kuva: Kaavio kaikista syistä aiheutuneista kuolemista suurimmissa kaupungeissa. Huippu saavutettiin loka- ja marraskuussa 1918
Kuva: Kaavio kaikista syistä aiheutuneista kuolemista suurimmissa kaupungeissa. Huippu saavutettiin loka- ja marraskuussa 1918
Jopa alueilla, joilla kuolleisuus oli alhainen, monet sairastuneet olivat työkyvyttömiä. Tämä häiritsi laajasti jokapäiväistä elämää. Jotkut yhteisöt sulkivat kaikki myymälät tai vaativat asiakkaita jättämään ja noutamaan tilauksensa myymälöiden ulkopuolelta.
Aikalaisraporttien mukaan terveydenhuollon työntekijät eivät voineet hoitaa sairaita ja kuolleiden hautaaminen viivästyi, koska sairaanhoitajista ja haudankaivajista suuri osa oli sairaana.
Joukkohautoja kaivettiin ja ruumiit haudattiin monin paikoin ilman arkkuja [216]. Alaskan alkuperäiskansojen asuttamalla Bristol Bayn alueella kuoli 40 prosenttia koko väestöstä. Jotkut kylät katosivat kokonaan [217].
Monet Tyynenmeren saarialueet kärsivät espanjantaudista ankarasti. Pandemia saavutti saaret hitaasti pandemiaan reagoineen Uuden-Seelannin satamista lähteneiden laivojen mukana. Uudesta-Seelannista espanjantauti levisi Tongaan, jossa se tappoi 8 % väestöstä, Nauruun (kuolleisuus16 %) ja Fidžiin, jossa kuoleisuus oli 5 %väestöstä (9 000 ihmistä) [218].
Pahimmin kärsi Länsi-Samoa (entinen Saksan Samoa), jonka Uusi-Seelanti oli miehittänyt vuonna 1914. Länsi-Samoan väestöstä 90 % sai tartunnan (30 % miehistä, 22 % naisista ja 10 % lapsista kuoli pandemiaan). Kuvernööri John Martin Poyer sen sijaan onnistui estämään pandemian leviämisen Amerikan Samoaan määräämällä saarron [218].
Espanjantauti levisi nopeimmin alkuperäiskansojen keskuudessa, koska alkuperäiskansojen vanhimmat kokoontuivat perinteitä kunnioittaen päälliköiden kuolinvuoteelle kuulemaan suullista perimätietoa ja oppimaan päälliköiden viisautta.Monet heimojen vanhimmat saivat tämän perinteen vuoksi tartunnan [219].
Iranissa kuolleisuus oli hyvin korkea: arvioiden mukaan 902 400–2 431 000 eli 8–22 prosenttia koko Iranin väestöstä kuoli pandemiaan [220] ja samanaikaiseen Persiassa vuosina 1917-1919 riehuneeseen nälänhätään. Irlannissa espanjantauti aiheutti pahimpien 12 kuukauden aikana kolmanneksen kaikista kuolemista [221][222].
Etelä-Afrikassa arvioidaan, että noin 300 000 ihmistä (6 % väestöstä) kuoli kuuden viikon aikana 1918. Hallituksen toimien uskotaan tahattomasti kiihdyttäneen viruksen leviämistä koko maassa [223]. Melkein neljännes Kimberleyn timanttikaivosten työläisistä kuoli [224].
Brittiläisessä Somalimaassa 7 % alkuperäisväestöstä kuoli [225]. Kuolonuhrien määrä johtui erittäin korkeasta (50 %:n) tartuntatasosta ja oireiden vakavuudesta. Monet kuolivat taudin käynnistämään sytokiinimyrskyyn [96].
Vähemmän kärsineet alueet
Tyynellämerellä Amerikan Samoa [226] ja Ranskan Uusi-Kaledonia [227] onnistuivat estämään influenssakuolemat tehokkaiden karanteenien avulla. Karanteenikauden päätyttyä epidemian puhkeaminen viivästyi vuoteen 1926 Amerikan Samoalla ja 1921 Uudessa-Kaledoniassa [228].
Amerikan Samoalla vähintään 25 % saaren asukkaista sai tartunnan ja 0,1 % kuoli. Uudessa-Kaledoniassa tauti levisi laajasti ja 0,1 % väestöstä kuoli [228].
Australia onnistui välttämään espanjantaudin kaksi ensimmäistä aaltoa karanteeneilla [172]. Islanti suojeli kolmasosaa väestöstään altistumiselta sulkemalla saaren päätien [172]. Pandemian loppuun mennessä eristetty Marajón saari Brasilian Amazonjoen suistossa säästyi tartunnoilta [229]. St. Helenalla ei rekisteröity kuolemantapauksia [230].
 Kuva: Japanilaiset naiset käyttävät valkoisia sideharsomaskia Espanjan flunssapandemian aikana Tokiossa, Japanissa, 1919
Kuva: Japanilaiset naiset käyttävät valkoisia sideharsomaskia Espanjan flunssapandemian aikana Tokiossa, Japanissa, 1919
Arviot Kiinan kuolonuhrien määrästä ovat vaihdelleet suuresti [231][96]. Vaihtelu johtuu sotapäällikköiden kauden keskitettyjen terveystietojen keruun puutteesta. Kiinassa saattoi olla suhteellisen lievä influenssakausi vuonna 1918 verrattuna muuhun maailmaan [137][139][232]. Toisaalta jotkut raportit viittaavat siihen, että vuoden 1918 influenssakuolleisuus oli oletettua korkeampi ainakin eräissä Kiinan osissa [215]. Näyttöä vakavasta koko Kiinaan vaikuttaneesta influenssasta ei kuitenkaan ole [233].
Ensimmäisen arvion Kiinan kuolonuhrien määrästä tekivät Patterson ja Pyle vuonna 1991. He arvioivat kuolonuhrien määräksi 5-9 miljoonaa. Pattersonin ja Pylen tutkimusta on kritisoitu myöhemmin puutteellisen metodologian vuoksi, Uuudemmissa tutkimuksissa on julkaistu arvioita merkittävästi alhaisemmasta kuolleisuudesta [135][234].Esimerkiksi Iijima arvioi kuolonuhrien määräksi 1–1,28 miljoonaa Kiinan satamakaupunkien tietojen perusteella [235]. Alhaisemmat kuolinluvut perustuvat havaintoihin matalasta kuolleisuudesta Kiinan satamakaupungiessa [231]. Jotkut sanomalehti- ja postiraportit sekä lähetyssaarnaajien lääkäreiden raportit viittaavat kuitenkin siihen, että espanjantauti tunkeutui Kiinaan ja että influenssa oli vakava ainakin Kiinan maaseudulla [215]. Vaikka Kiinan sisämaasta puuttuu potilastietoja, laajat lääketieteelliset tiedot tallentiin Kiinan satamakaupungeissa, kuten brittien hallitsemissa Hongkongissa, Kantonissa, Pekingissä, Harbinissa ja Shanghaissa. Tiedot keräsi Kiinan meritullipalvelu, jossa työskenteli paljon ei-kiinalaisia, kuten brittejä, ranskalaisia ja muita eurooppalaisia siirtomaavirkamiehiä [236].
Kiinan satamakaupunkien tiedot vahvistavat alhaisemman pandemiakuolleisuuden verrattuna moniin muihin Aasian kaupunkeihin [236]. Esimerkiksi Britannian viranomaiset raportoivat Hongkongissa ja Kantonissa influenssakuolleisuusasteeksi 0,25 % ja 0,32 %, jotka ovat paljon matalammat kuin muissa Aasian kaupungeissa; esimerkiksi Kalkutassa ja Bombayssa espanjantauti oli selvästi tuhoisampi [236]. Shanghaissa (jonka väkiluku oli yli 2 miljoonaa vuonna 1918) kiinalaisilla kirjattiin vain 266 influenssakuolemaa vuonna 1918 [236].
Kiinan kaupungeista tallennetuista laajoista tiedoista ekstrapoloituna espanjantaudin arvioitu kuolleisuusaste koko Kiinassa vuonna 1918 oli todennäköisesti alle 1 %, eli paljon pienempi kuin maailman keskiarvo (joka oli noin 3–5 %) [236].
Japanissa ja Taiwanissa espanjantaudin kuolleisuusaste oli noin 0,45 % ja 0,69 %, mikä on korkeampi kuin Kiinan satamakaupunkien, kuten Hongkongin (0,25 %), Kantonin (0,32 %) ja Shanghain kuolleisuusaste [236].
On kuitenkin huomattava, että influenssakuolleisuus Hongkongissa ja Kantonissa on alirekisteröity, koska vain siirtokuntien sairaaloissa tapahtuneet kuolemat rekisteröitiin [236]. Shanghaissa nämä tilastot rajoittuvat siihen kaupungin alueeseen, joka oli Shanghain kansainvälisen järjestelyn terveysosaston hallinnassa, ja todellinen kuolonuhrien määrä Shanghaissa oli paljon korkeampi [236]. Kiinan sisämaasta saadut potilastiedot osoittavat, että maaseutuyhteisöiss kuolleisuus oli selvästi korkeampi kuin kaupungeissa [237].
Tutkimus Houlun läänistä Hebein maakunnasta osoitti, että sairastuneiden kuolleisuusaste oli 9,77 %. Läänin väestöstä 0,79 % kuoli influenssaan loka- ja marraskuussa 1918 [238].
Kuoleman kaavat
 Kuva: Kasvonaamiota käyttävä hoitaja Washington, DC
Kuva: Kasvonaamiota käyttävä hoitaja Washington, DC
Pandemia tappoi enimmäkseen nuoria aikuisia. Vuosina 1918–1919 99 % pandemian aiheuttamista influenssakuolemista Yhdysvalloissa tapahtui alle 65-vuotiailla. Lähes puolet espanjantautiin kuolleista oli 20–40-vuotiaita nuoria aikuisia. Vuoteen 1920 mennessä alle 65-vuotiaiden kuolleisuus laski kuusinkertaisesta puoleen suhteessa yli 65-vuotiaiden kuolleisuuteen, mutta 92 % kuolemista tapahtui edelleen alle 65-vuotiailla [239].
Tämä oli epätavallista, koska influenssa on tyypillisesti tappavin heikoille yksilöille, kuten alle kaksivuotiaille vauvoille, yli 70-vuotiaille ja immuunipuutteisille. Vuonna 1918 ikääntyneillä saattoi olla osittainen immuniteetti espanjantaudin vakavalle muodolle, joka johtui altistumisesta vuosien 1889–1890 influenssapandemialle, eli ”venäläiselle influenssalle” [240].
Historioitsija John M. Barryn mukaan espanjantautiin ”todennäköisimmin kuolevat” olivat raskaana olevat naiset. Barry raportoi, että 13 tutkimuksessa pandemian aikana sairaalahoidossa olevien naisten kuolleisuus vaihteli 23 prosentista 71 prosenttiin [241]. Raskaana olevista naisista, jotka selvisivät synnytyksestä, yli neljäsosa (26 %) menetti lapsensa [242].
Toinen espanjantaudin omituisuus oli, että epidemia levisi pohjoisella pallonpuoliskolla erityisesti kesällä ja syksyllä; influenssa on yleensä pahempi talvella [243].
Taudin kuolleisuuteen liittyi myös kiinnostavia maantieteellisiä eroja. Joissakin osissa Aasiaa kuolleisuus oli 30 kertaa korkeampi kuin osissa Eurooppaa. Afrikassa ja Aasiassa kuolleisuus oli yleisesti korkeampi, kun taas Euroopassa ja Pohjois-Amerikassa [244]. Myös maanosien sisällä oli suuria eroja: Unkarissa ja Espanjassa kuolleisuus oli kolme kertaa korkeampi kuin Tanskassa, Saharan eteläpuolisessa Afrikassa kuolleisuus oli Pohjois-Afrikkaan verrattuna 2-3 kertaa suurempi ja jopa kymmenen kertaa korkeampi äärimmäisyyksien välillä Aasiassa [244].
Kaupungit kärsivät espanjantaudista yleensä pahemmin kuin maaseutu [244]. Kaupunkien välillä oli kuitenkin huomattavia eroja, mikä saattoi selittyä kaupunkilaisten taudin lievälle ensimmäiselle aallolle altistumisen tuottamasta immuniteetista sekä sosiaalisten etäisyyksien käyttöönotosta [245].
Toinen merkittävä havainto espanjantautikuolleisuudesta oli yhteiskuntaluokkien väliset erot. Oslossa kuolleisuus korreloi käänteisesti asunnon koon kanssa; pienemmissä asunnoissa eläviä köyhiä kuoli enemmän kuin varakkaampia ja väljemmin asuvia [246]. Sosiaalinen asema heijastui myös maahanmuuttajayhteisöjen korkeampana kuolleisuutena. Esimerkiksi hiljattain Yhdysvaltoihin muuttaneet amerikanitalialaiset kuolivat espanjantautiin lähes kaksi kertaa todennäköisemmin kuin amerikkalaiset keskimäärin [244]. Erot johtuivat huonommasta ruokavaliosta, ahtaista elinoloista ja terveydenhuoltoon pääsyn ongelmista [244].
Paradoksaalista kyllä, afroamerikkalaiset säästyivät pandemialta suhteellisen hyvin [244]. Influenssaan kuoli enemmän miehiä kuin naisia, koska miehet menivät todennäköisemmin ulos ja altistuivat virukselle mm. työyhteisössä; naisten sosiaalinen elämä oli rajoitetumpaa ja kotiin sidotumpaa [245]. Samasta syystä miehet sairastivat todennäköisemmin tuberkuloosia, mikä heikensi toipumismahdollisuuksia espanjantaudista [245]. Intiassa tilanne oli päinvastainen ehkä siksi, että intialaisia naisia ravittiin huonommalla ravinnolla ja heidän odotettiin hoitavan sairaita [245].
He et al. (2011) käytti mekanistista mallintamista tutkiessaan vuoden 1918 influenssapandemian kolmea aaltoa. He tutki taustalla vaikuttavia tekijöitä, jotka selittävät vaihtelua epidemian ajallisissa malleissa ja niiden korrelaatiota kuolleisuuden ja sairastuvuuden malleihin. Analyysi viittaa siihen, että taudin leviämisnopeuden ajalliset vaihtelut tarjoavat parhaan selityksen, ja näiden kolmen aallon synnyttämiseen vaadittava transmissio on biologisesti uskottavien arvojen sisällä [247].
Toisessa tutkimuksessa He et al. (2013) käytti yksinkertaista epidemiamallia, joka sisälsi kolme muuttujaa päätelläkseen syyn vuoden 1918 influenssapandemian kolmelle aallolle. Näitä tekijöitä olivat koulujen avaaminen ja sulkeminen, lämpötilan muutokset koko epidemian aikana ja ihmisten käyttäytymisen muutokset tautiaaltojen aikana. Mallinnustulokset osoittivat, että kaikki kolme tekijää olivat tärkeitä, mutta ihmisen käyttäytymisvasteet osoittivat merkittävimmät vaikutukset [248].
Vaikutukset
Ensimmäinen maailmansota
Akateemikko Andrew Price-Smith väittää, että virus kallisti voimatasapainoa sodan viimeisinä päivinä liittoutuneiden eduksi. Hän esitttää perusteluksi tietoja siitä, että virusaallot osuivat keskusvaltioihin ennen liittoutuneita ja että sekä sairastuvuus että kuolleisuus Saksassa ja Itävallassa olivat huomattavasti korkeammat kuin Britanniassa ja Ranskassa [129].
Vuoden 2006 Lancet-tutkimus vahvistaa korkeamman kuolleisuusasteen Saksassa (0,76 %) ja Itävallassa (1,61 %) verrattuna Isoon-Britanniaan (0,34 %) ja Ranskaan (0,75 %) [194].
Kenneth Kahn Oxfordin yliopiston tietotekniikkapalveluista kirjoittaa, että ”Monet tutkijat ovat ehdottaneet, että sodan olosuhteet auttoivat merkittävästi taudin leviämistä. Ja toiset ovat väittäneet, että pandemia vaikutti sodan (ja myöhemmän rauhansopimuksen) kulkuun.” Kahn on kehittänyt mallin, jota voidaan käyttää kotitietokoneissa näiden teorioiden testaamiseen [249].
Talous
 Kuva: Albertan maakuntahallituksen terveysjuliste
Kuva: Albertan maakuntahallituksen terveysjuliste
Monet viihde- ja palveluteollisuuden yritykset menettivät tuloja, kun taas terveydenhuoltoala raportoi voitoista [250]. Historioitsija Nancy Bristow kertoo, että pandemia ja korkeakoulutettujen naisten määrän lisääntymiseen selittää naisten menestystä hoitotyön aloilla.Tämä johtui osittain siitä, että lääkärit, jotka olivat pääosin miehiä, kantoivat yleisön silmissä vastuun epidemiasta, koska he eivät pystyneet hillitsemään ja ehkäisemään taudin leviämistä.
Taudista ei syytetty hoitohenkilöstöä, joka oli pääosin naisia. Sen sijaan naisia kiitettiin ja arvostettiin potilashoidon onnistumisesta [251].
Vuonna 2020 tehdyssä tutkimuksessa havaittiin, että Yhdysvaltojen kaupungit, jotka panivat täytäntöön varhaisia ja laajoja ei-lääketieteellisiä toimenpiteitä (karanteeni jne.), eivät kärsineet ylimääräisistä haitallisista taloudellisista vaikutuksista näiden toimenpiteiden toteuttamisen vuoksi [252], toisin kuin kaupungit, jotka panivat toimenpiteitä täytäntöön myöhässä tai eivät ollenkaan [252] 253][254].
Pitkäaikaisvaikutukset
Journal of Political Economy -lehdessä vuonna 2006 julkaistussa tutkimuksessa kerrottiin, että pandemia-aikana odottavien naisten synnyttämien lasten koulutustaso oli verrokkikohortteja heikompi ja lasten fyysiset vammat (disabiliteetti) yleisempiä. Lisäksi tulevat ansiotulot ja sosioekonominen asema olivat pandemian aikaan syntyneillä muihin syntymäkohortteihin nähden verrokkeja heikommat [255] ]. Vuoden 2018 tutkimuksessa havaittiin, että pandemia alensi väestön koulutustasoa [256].
Espanjantauti assosioituu 1920-luvun enkefalitis lethargican puhkeamiseen [257]. Taudista toipuneiden kuolleisuusriski muihin infektioihin ja sairauksiin kasvoi. Jotkut eloonjääneet eivät koskaan toipuneet espanjantaudin aiheuttamista fysiologisista oireista [258].
Espanjantaudin perintö – unohdettu pandemia
Huolimatta espanjantautipandemian aiheuttamista korkeasta sairastuvuus- ja kuolleisuusluvuista, espanjantauti hiipui yleisestä tietoisuudesta vuosikymmeniksi, kunnes lintuinfluenssasta ja muista pandemioista tuli uutisia 1990- ja 2000-luvuilla [259].
Jotkut historioitsijat nimittävät espanjantautipandemiaa ”unohdetuksi pandemiaksi” [121]. Koronapandemia on viimeisten 1-2 vuoden aikana palauttanut espanjantautipandemian laajemman yleisön tietoon.
 Kuva: 1918 influenssahautoja, Auckland, Uusi-Seelanti
Kuva: 1918 influenssahautoja, Auckland, Uusi-Seelanti
On monia teorioita siitä, miksi espanjantauti ”unohdettiin” vuosikymmeniksi. Pandemia tappoi suurimman osan uhreistaan Yhdysvalloissa alle yhdeksässä kuukaudessa, minkä vuoksi taudin medianäkyvyys jäi lyhytaikaiseksi. Väestö tunsi pandeemisten tautien mallit 1800-luvun lopulla ja 1900-luvun alussa: lavantauti, keltakuume, kurkkumätä ja kolera esiintyivät lähes samaan aikaan.
Nämä epidemiat todennäköisesti vähensivät influenssapandemian saamaa huomiota [260].
Joillain alueilla influenssasta ei raportoitu lainkaan. Ainoa maininta influenssapandemiasta saattoi olla lääkemainokset, joissa myytiin influenssan ”parantavia” lääkkeitä [261].
Espanjantaudin aikana media keskittyi ensimmäiseen maailmansotaan ja sen uhreihin [262]. Yksi selitys taudin huonolle tuntemukselle liittyy ikäryhmään: suurin osa sekä sotaan ja espanjantautiin kuolleista oli nuoria aikuisia. Nuorten aikuisten sotaan liittyvien kuolemantapausten suuri määrä on voinut varjostaa espanjantaudin aiheuttamia kuolemia [239].
Euroopassa, missä sodan uhrimäärät olivat korkeat, influenssapandemialla ei ole ollut suurta psykologista vaikutusta ihmisiin. Pandemia on saattanut tuntua sodan tragedioiden jatkeelta [239]. Sodan kesto vei huomion pandemialta: sodan oli alun perin odotettu päättyvän nopeasti, mutta se kesti neljä vuotta.
Espanjantaudin vertailu muihin pandemioihin
Espanjantauti tappoi paljon pienemmän osan maailman väestöstä kuin musta surma (rutto 1346-1353), joka kesti vuosia [264].
Yhä jatkuva COVID-19 pandemia on virallisten lukujen mukaan sairastuttanut13 helmikuuta 2022 mennessä 410 273 673 ihmistä. Koronaan on vahvistettu kuolleen 5 833 027 ihmistä, mutta koska kaikissa maissa tartuntoja ja koronaankuolleita ei rekisteröidä kovinkaan tarkasti, todellisen kuolleisuuden uskotaan olevan jopa neljä kertaa virallisia lukuja korkeampi [265].
Vakavimmat influenssapandemiat [266][267]
| Nimi | Aika | Pop. | Tyyppi | Tarttuvuus [268] | Tartunnat | Kuolemat maailma | Kuolleisuusaste | Pandemia vakavuus |
|---|---|---|---|---|---|---|---|---|
| 1889–90 flu pandemic[269] | 1889–90 | 1.53 miljardia | Likely H3N8 or H2N2 | 2.10 (IQR, 1.9–2.4)[269] | 20–60%[269] (300–900 million) | 1 million | 0.10–0.28%[269] | 2 |
| Espanjantauti[270] | 1918–20 | 1.80 miljardia | H1N1 | 1.80 (IQR, 1.47–2.27) | 33% (500 million)[271] or >56% (>1 miljardia)[272] | 17[273]–100[274][275] million | 2–3%,[272] or ~4%, or ~10%[276] | 5 |
| Asian flu | 1957–58 | 2.90 miljardia | H2N2 | 1.65 (IQR, 1.53–1.70) | >17% (>500 million)[272] | 1–4 million[272] | <0.2%[272] | 2 |
| Hong Kong flu | 1968–69 | 3.53 miljardia | H3N2 | 1.80 (IQR, 1.56–1.85) | >14% (>500 million)[272] | 1–4 million[272] | <0.2%[272][277] | 2 |
| 1977 Russian flu | 1977–79 | 4.21 miljardia | H1N1 | ? | ? | 0.7 million[278] | ? | ? |
| 2009 swine flu pandemic[279][280] | 2009–10 | 6.85 miljardia | H1N1/09 | 1.46 (IQR, 1.30–1.70) | 11–21% (0.7–1.4 miljardia)[281] | 151,700–575,400[282] | 0.01%[283][284] | 1 |
| Typical seasonal flu[t 1] | Every year | 7.75 miljardia | A/H3N2, A/H1N1, B, … | 1.28 (IQR, 1.19–1.37) | 5–15% (340 million – 1 miljardia)[285] 3–11% or 5–20%[286][287] (240 million – 1.6 billion) |
290,000–650,000/year[288] | <0.1%[289] | 1 |
|
|
||||||||
Tutkimus
 Kuva: Elektronimikroskooppikuva, joka näyttää reproduktiona luodut 1918 influenssavirionit
Kuva: Elektronimikroskooppikuva, joka näyttää reproduktiona luodut 1918 influenssavirionit
Espanjantautipandemian alkuperä sekä ihmisten ja sikojen lähes samanaikaisten epidemioiden välinen suhde ovat kiistanalaisia.
Yksi hypoteesi on, että espanjantautia aiheuttava viruskanta sai alkunsa Fort Rileystä, Kansasista, varuskunnan ravinnoksi kasvattamien siipikarjan ja sikojen viruksista. Tämän hypoteesin mukaan sotilaat, jotka lähetettiin Fort Rileystä ympäri maailmaa, levittivät tautia [290].
Samankaltaisuudet ihmisten levittämän viruksen ja lintuinfluenssavirusten rekonstruoinnin välillä yhdistettynä pandemiaan, joka edelsi ensimmäisiä sikainfluenssaraportteja, sai tutkijat päättelemään, että influenssavirus hyppäsi suoraan linnuista ihmisiin ja tauti levisi sikoihin ihmisiltä [291] [292].
Kaikki eivät ole alkuperästä samaa mieltä [293]. Uudemmat tutkimukset vahvistavat, että kanta on saattanut olla peräisin muusta nisäkäslajista, kuin ihmisestä [294]. Arvioitu päivämäärä viruskannan ilmestymiselle nisäkäsisäntiin on asetettu ajanjaksolle 1882–1913 [295].
Alkuperäinen virus jakautui vuosien 1913–1915 aikana kahteen kladiin (biologiseen ryhmään), mikä johti sika- ja ihmisen H1N1-influenssalinjaan.
Ihmisten välillä leviävien viruskantojen viimeinen yhteinen esi-isä on ajoitettu helmikuun 1917 ja huhtikuun 1918 välille. Koska lintuinfluenssavirukset tarttuvat herkemmin sikoihin, kuin ihmisiin, niitä ehdotettiin viruksen alkuperäisiksi vastaanottajiksi, jotka välittivät viruksen ihmisiin joskus vuosina 1913-1918.
 Kuva: Terrence Tumpey tutkii sairauksien torjunta- ja ehkäisykeskuksessa (CDC) espanjantaudin rekonstruoitua versiota.
Kuva: Terrence Tumpey tutkii sairauksien torjunta- ja ehkäisykeskuksessa (CDC) espanjantaudin rekonstruoitua versiota.
Espanjantaudin viruskannan (lintuinfluenssan H1N1 alatyyppi) rekonstruktio tehtiin yhteistyössä Armed Forces Institute of Pathology, USDA ARS Southeast Poultry Research Laboratory ja Mount Sinai School of Medicine New Yorkissa yhteistyönä.
Tutkijat julkaisivat 5. lokakuuta 2005 tiedotteen, että he olivat onnistunut määrittämään viruksen geneettisen sekvenssin tutkimalla patologi Johan Hultinin espanjantautiin kuolleelta Alaskan ikiroutaan haudatulta inuiittinaiselta keräämiä kudosnäytteitä, sekä näytteitä, joita oli säilytetty amerikkalaisista sotilaista [296][297][298].
18. tammikuuta 2007 Kobasa et al. raportoi, että apinoilla, joihin oli infektoitu espanjantautia aiheuttaneen viruksen reproduktio, havaittiin klassisia vuoden 1918 pandemian oireita ja ne kuolivat sytokiinimyrskyyn, eli immuunijärjestelmän ylireagointiin [299]. Sytokiinimyrsky voi selittää, miksi espanjantaudilla oli niin raju vaikutus nuorempiin ja terveempiin ihmisiin. Vahvempi immuunijärjestelmä ylireagoi herkemmin [300].
16. syyskuuta 2008 brittiläisen poliitikon ja diplomaatin Sir Mark Sykesin ruumis kaivettiin haudasta influenssaviruksen RNA:n tutkimiseksi ja nykyaikaisen H5N1-lintuinfluenssan geneettisen rakenteen ymmärtämiseksi. Sykes oli haudattu vuonna 1919 lyijyarkkuun, jonka tutkijat toivoivat säilyttäneen viruksen [301]. Arkun todettiin haljenneen ja ruumis oli pahasti hajonnut; siitä huolimatta Sykesin ruumiista otettiin keuhko- ja aivokudosnäytteitä [302]. Joulukuussa 2008 Wisconsinin yliopiston Yoshihiro Kawaokan tutkimus havaitsi kolmen geenin (PA, PB1 ja PB2) ja espanjantautiäytteistä peräisin olevan nukleoproteiinin vaikuttavan influenssaviruksen kykyyn tunkeutua keuhkoihin ja aiheuttaa keuhkokuume. Tietty geneettinen yhdistelmä ja H1N1 viruksen alatyypin nukleoproteiini laukaisi espanjantautiin liittyviä oireita eläinkokeissa [303].
Kesäkuussa 2010 Mount Sinai School of Medicine -koulun työryhmä raportoi, että vuoden 2009 influenssapandemiarokote tarjosi jonkin verran ristisuojaa espanjalaisinfluenssapandemiakantaa vastaan [304]. Yksi harvoista asioista, jotka tiedettiin varmaksi influenssasta vuonna 1918 ja muutaman vuoden ajan sen jälkeen, oli, että se oli laboratoriota lukuun ottamatta yksinomaan ihmisten välillä leviävä sairaus [305].
Vuonna 2013 AIR Worldwide Research and Modeling Group kuvaili vuoden 1918 pandemiaa ja arvioi samanlaisen nykyään esiintyvän pandemian vaikutuksia käyttämällä AIR Pandemic Flu Model -mallia. Mallissa ”nykyajan ”espanjalainen flunssa” aiheuttaisi 15,3–27,8 miljardin dollarin edestä taloudellisia vahinkoja pelkästään Yhdysvalloissa ja 188 000–337 000 kuolemaa Yhdysvalloissa [306].
Vuonna 2018 Michael Worobey, Arizonan yliopiston evoluutiobiologian professori, kertoi saaneensa William Rollandin ensimmäisen maailmansodan aikana keräämät kudosnäytteet. William Rolland oli lääkäri ja Britannian armeijan patologi, joka raportoi mahdollisesti viruksen aiheuttamasta hengitystiesairaudesta Lancetiin vuonna 1917 [307]. Rollandin viittaama hengitystiesairaus oli alkanut vuonna 1916 Étaplesissa, Ranskassa [308][309]. Worobey onnistui jäljittämään Rollandin perheenjäsenet, jotka olivat säilyttäneet tämän keräämät näytediat. Worobey poimi Rollandin näytteistä kudosta selvittääkseen enemmän patogeenin alkuperästä [28].
Vuonna 2021 tehdyssä tutkimuksessa [310] käytettiin virussekvenssiä [292] hemagglutiniini-antigeenin (HA) saamiseksi ja adaptiivisen immuniteetin havainnoimiseksi 32 vuoden 1918 influenssapandemiasta selvinneellä. Kaikilla heillä oli seroreaktiivisuutta ja 7 kahdeksasta muusta testatusta esitti immunologisen muistin B-solujen pystyvän tuottamaan vasta-aineita, jotka sitoutuivat HA-antigeeniin, mikä vahvisti immunologisen muistin toimivan jopa vuosikymmeniä.
Sukupuolierot kuolleisuudessa
Espanjantautipandemian korkea kuolleisuus on yksi tekijä, joka erottaa espanjantaudin muista influenssa-aalloista. Toinen tekijä oli miesten korkeampi kuolleisuus verrattuna naisiin. Miehillä, joilla oli jokin perussairaus, oli huomattavasti suurempi riski kuolla espanjantautiin. Tuberkuloosi oli yksi tappavimmista taudeista 1900-luvulla. Espanjantaudin leviämisen seurauksena miesten tuberkuloositapaukset vähenivät. Monet tutkijat ovat havainneet, että tuberkuloosi lisäsi miesten influenssakuolleisuutta ja lyhensi tuberkuloosia sairastavien eliniänodotetta. 1900-luvulla tuberkuloosi oli yleisempää miehillä kuin naisilla, mutta tutkimukset osoittavat, että influenssan leviämisen myötä naisten tuberkuloosikuolleisuus lisääntyi merkittävästi ja laskisi edelleen pandemian jälkeiseen aikaan asti [311].
Erityisen korkea kuolleisuus oli 20–35-vuotiailla. Ainoa tähän verrattavissa oleva sairaus oli musta surma, paiserutto 1300-luvulla.
Tutkimukset ovat osoittaneet, tuberkuloosi ja influenssa olivat samanaikaisia tauteja. Influenssaan kuolevien miesten iät vahvistavat, että tuberkuloosi oli yksi erityisesti miesten kuolleisuutta lisäävä tekijä. Koska miehillä esiintyi tuberkuloosia pandemian aikana, miesten kuolleisuus espanjantautiin lisääntyi. Miesten elinajanodote putosi pandemian aikana, mutta nousi kaksi vuotta pandemian jälkeen [312].
Newfoundland
Yksi syy espanjantaudin leviämiseen oli sosiaalinen käyttäytyminen. Miehillä oli enemmän sosiaalista elämää ja he liikkuivat enemmän kuin naiset työnsä vuoksi. Vaikka miesten kuolleisuus oli korkeampi, kullakin alueella saatiin erilaisia tuloksia esimerkiksi ravitsemuksen puutteen vuoksi.
Newfoundlandissa pandemian leviäminen oli erittäin vaihtelevaa. Influenssa ei tehnyt eroa siitä, kuka sai tartunnan, vaan sairastumisalttiuteen vaikutti ihmisten sosioekonominen asema. Vaikka sosiaalinen vaihtelevuus mahdollisti taudin nopean leviämisen maantieteellisesti, se levisi nopeammin ja vaikutti miehiin enemmän kuin naisiin työn ja sosiaalisten kontaktien vuoksi.
Newfoundlandin yleisin kuolinsyy ennen pandemiaa oli tuberkuloosi. Tämän tiedetään olevan vakava taustasairaus ja espanjantautikuolleisuutta lisännyt riskitekijä.
Newfoundlandissa oli monenlaista työvoimaa. Miehillä ja naisilla oli erilaisia ammatteja, joihin liittyi keskinäistä vuorovaikutusta. Kalastuksella oli tärkeä rooli taloudessa. Miehet liikkuivat enemmän kuin naiset ja heillä oli enemmän kontakteja muihin ihmisiin ja maailman osiin.
Pandemian leviämisen tiedetään alkaneen keväällä 1918, mutta Newfoundland koki tappavan aallon vasta kesä- tai heinäkuussa, mikä on linjassa kalastuksen suuren työllisyyden kanssa. Suurin osa miehistä työskenteli rannikolla kesän aikana ja oli tyypillistä, että kokonaiset perheet muuttivat kesäisin Newfoundlandiin töihin.
Mutta pandemian ensimmäisen, toisen ja kolmannen aallon aikana kuolleisuus muuttui. Ensimmäisen aallon aikana miesten kuolleisuus oli korkeampi, mutta naisten kuolleisuus kasvoi ja oli korkeampi toisen ja kolmannen aallon aikana. Naisväestö oli suurempi tietyillä Newfoundlandin alueilla, ja siksi sillä oli suurempi vaikutus kuolleisuuteen [313].
Espanjantauti kanadalaisilla sotilailla
Arkistojen mukaan pandemian ensimmäisen aallon aikana kuoli eniten 20-vuotiaita nuoria miehiä, mikä heijastaa sotaan värvättyjen ikää. Nuorten miesten liikkuvuus vuonna 1918 yhdistettiin influenssan leviämiseen ja epidemian suurimpaan aaltoon.
Vuoden 1917 lopulla ja vuoden 1918 aikana tuhansista miehistä koostuvat joukot kokoontuivat Halifaxin satamaan ennen kuljetusta Eurooppaan. Kaikki sairaat sotilaat, jotka eivät voineet lähteä, rekisteröitiin Halifaxin väestöön. Tämä lisäsi miesten influenssatapausten määrää sodan aikana.
Määrittääkseen kuolinsyyn pandemian aikana, sotatieteilijät käyttivät 1917-1918 kuolleisuustilastoja ja Commonwealth War Graves Commissionia (CWGC), joka raportoi alle 2 miljoonan miehen ja naisen kuolleen sotien aikana.
Sotilaiden määrä ja kuljetukset Yhdysvaltojen ja Kanadan välillä todennäköisesti vaikuttivat merkittävästi pandemian leviämiseen.[314]
 Kuva: H1N1 virus
Kuva: H1N1 virus
Bibliografia
- Antonovics J, Hood ME, Baker CH (April 2006). ”Molecular virology: was the 1918 flu avian in origin?”. Nature. 440 (7088): E9, discussion E9–10. Bibcode:2006Natur.440E…9A. doi:10.1038/nature04824. PMID 16641950. S2CID 4382489.
- Afkhami A (2003). ”Compromised constitutions: the Iranian experience with the 1918 influenza pandemic”. Bulletin of the History of Medicine. 77 (2): 367–92. doi:10.1353/bhm.2003.0049. PMID 12955964. S2CID 37523219. Archived from the original on 31 March 2020. Retrieved 3 October 2017. – Open access material by the Psychiatry and Behavioral Sciences at Health Sciences Research Commons.
- Afkhami A (29 March 2012) [15 December 2004]. ”Influenza”. In Yarshater E (ed.). Encyclopædia Iranica. Fasc. 2. XIII (Online ed.). New York City: Bibliotheca Persica Press. pp. 140–43. Archived from the original on 27 November 2020. Retrieved 3 October 2017.
- Barry JM (2004). The Great Influenza: The Epic Story of the Greatest Plague in History. Viking Penguin. ISBN 978-0-670-89473-4.
- Barry JM (January 2004b). ”The site of origin of the 1918 influenza pandemic and its public health implications”. Journal of Translational Medicine. 2 (1): 3. doi:10.1186/1479-5876-2-3. PMC 340389. PMID 14733617.
- Benedict ML, Braithwaite M (2000). ”The Year of the Killer Flu”. In the Face of Disaster: True Stories of Canadian Heroes from the Archives of Maclean’s. New York: Viking. p. 38. ISBN 978-0-670-88883-2.
- Billings M (1997). ”The 1918 Influenza Pandemic”. Virology at Stanford University. Archived from the original on 4 May 2009. Retrieved 1 May 2009.
- Bristow NK (2012). American Pandemic: The Lost Worlds of the 1918 Influenza Epidemic. New York: OUP. ISBN 978-0-19-023855-1. OCLC 1231722653.
- Chandra S, Kuljanin G, Wray J (August 2012). ”Mortality from the influenza pandemic of 1918–1919: the case of India”. Demography. 49 (3): 857–65. doi:10.1007/s13524-012-0116-x. PMID 22661303. S2CID 39247719.
- Crosby AW (1976). Epidemic and Peace, 1918. Westport, CT: Greenwood Press. ISBN 978-0-8371-8376-3.
- Crosby AW (2003). America’s Forgotten Pandemic: The Influenza of 1918 (2nd ed.). Cambridge University Press. ISBN 978-0-521-54175-6. Archived from the original on 3 June 2020. Retrieved 3 May 2020 – via Google Books.
- Davis RA (2013). The Spanish Flu: Narrative and Cultural Identity in Spain, 1918. Palgrave Macmillan. ISBN 978-1-137-33921-8. Archived from the original on 27 July 2021. Retrieved 9 November 2020 – via Google Books.
- Denoon D (2004). ”New Economic Orders: Land, Labour and Dependency”. In Denoon D (ed.). The Cambridge History of the Pacific Islanders. CUP. p. 247. ISBN 978-0-521-00354-4.
- Dionne, K.Y; Turkmen, FF (2020). ”The Politics of Pandemic Othering: Putting COVID-19 in Global and Historical Context”. International Organization. 74 (S1): E213–E230. doi:10.1017/S0020818320000405. S2CID 230561266.
- dos Reis M, Hay AJ, Goldstein RA (October 2009). ”Using non-homogeneous models of nucleotide substitution to identify host shift events: application to the origin of the 1918 ’Spanish’ influenza pandemic virus”. Journal of Molecular Evolution. Springer Science and Business Media LLC. 69 (4): 333–45. Bibcode:2009JMolE..69..333D. doi:10.1007/s00239-009-9282-x. PMC 2772961. PMID 19787384.
- Ewald PW (1994). Evolution of infectious disease. OUP. ISBN 978-0-19-506058-4.
- Gagnon, A; Miller, MS; Hallman, SA; Bourbeau, R; Herring, D.A; Earn, DJ; Madrenas, J (2013). ”Age-specific mortality during the 1918 influenza pandemic: unravelling the mystery of high young adult mortality”. PLOS ONE. 8 (8): e69586. Bibcode:2013PLoSO…869586G. doi:10.1371/journal.pone.0069586. PMC 3734171. PMID 23940526.
- Fox M (29 December 2008). ”Researchers unlock secrets of 1918 flu pandemic”. Reuters. Archived from the original on 9 November 2020. Retrieved 2 September 2009.
- Fox M (16 June 2010). ”Swine flu shot protects against 1918 flu: study”. Reuters. Archived from the original on 18 June 2010. Retrieved 30 June 2017.
- Galvin J (31 July 2007). ”Spanish Flu Pandemic: 1918”. Popular Mechanics. Archived from the original on 20 September 2011. Retrieved 2 October 2011.
- Garret TA (2007). Economic Effects of the 1918 Influenza Pandemic: Implications for a Modern-day Pandemic (PDF). Archived (PDF) from the original on 22 September 2020. Retrieved 22 November 2016.
- Gladwell M (29 September 1997). ”The Dead Zone”. The New Yorker. Archived from the original on 15 December 2020. Retrieved 16 March 2020.
- Hays JN (1998). The Burdens of Disease: Epidemics and Human Response in Western History. p. 274. ISBN 978-0-8135-2528-0. Archived from the original on 27 July 2021. Retrieved 3 May 2020 – via Google Books.
- He D, Dushoff J, Day T, Ma J, Earn DJ (2011). ”Mechanistic modelling of the three waves of the 1918 influenza pandemic”. Theoretical Ecology. 4 (2): 283–88. doi:10.1007/s12080-011-0123-3. ISSN 1874-1738. S2CID 2010776.
- He D, Dushoff J, Day T, Ma J, Earn DJ (September 2013). ”Inferring the causes of the three waves of the 1918 influenza pandemic in England and Wales”. Proceedings. Biological Sciences. 280 (1766): 20131345. doi:10.1098/rspb.2013.1345. PMC 3730600. PMID 23843396.
- Honigsbaum M (2008). Living with Enza: The Forgotten Story of Britain and the Great Flu Pandemic of 1918. Palgrave Macmillan. ISBN 978-0-230-21774-4.
- Humphries MO (2014). ”Paths of Infection: The First World War and the Origins of the 1918 Influenza Pandemic”. War in History. 21 (1): 55–81. doi:10.1177/0968344513504525. S2CID 159563767.
- Johnson NP, Mueller J (2002). ”Updating the accounts: global mortality of the 1918–1920 ”Spanish” influenza pandemic”. Bulletin of the History of Medicine. 76 (1): 105–15. doi:10.1353/bhm.2002.0022. PMID 11875246. S2CID 22974230.
- Knobler S, Mack A, Mahmoud A, Lemon S, eds. (2005). ”1: The Story of Influenza”. The Threat of Pandemic Influenza: Are We Ready? Workshop Summary (2005). Washington, DC: The National Academies Press. pp. 60–61. Archived from the original on 23 December 2020. Retrieved 20 August 2019.
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. (January 2007). ”Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus”. Nature. 445 (7125): 319–23. Bibcode:2007Natur.445..319K. doi:10.1038/nature05495. PMID 17230189. S2CID 4431644.
- Kohn GC (2007). Encyclopedia of plague and pestilence: from ancient times to the present (3rd ed.). Infobase Publishing. p. 363. ISBN 978-0-8160-6935-4. Archived from the original on 1 January 2016. Retrieved 29 October 2015 – via Google Books.
- Madhav N (February 2013). ”Modeling a Modern Day Spanish Flu Pandemic” (PDF). AIR Worldwide. Archived from the original on 26 February 2020. Retrieved 5 May 2020.
- More; Loveluck; Clifford; Handley; Korotkikh; Kurbatov; McCormick; Mayewski (September 2020). ”The Impact of a Six-Year Climate Anomaly on the ”Spanish Flu” Pandemic and WWI”. GeoHealth. 4 (9): e2020GH000277. doi:10.1029/2020GH000277. PMC 7513628. PMID 33005839.
- Morrisey CR (1986). ”The Influenza Epidemic of 1918”. Navy Medicine. 77 (3): 11–17.
- Noymer A, Carreon D, Johnson N (April 2010). ”Questioning the salicylates and influenza pandemic mortality hypothesis in 1918–1919”. Clinical Infectious Diseases. 50 (8): 1203, author reply 1203. doi:10.1086/651472. PMID 20233050.
- Pankhurst R (1991). An Introduction to the Medical History of Ethiopia. Trenton: Red Sea Press. ISBN 978-0-932415-45-5.
- Patterson KD, Pyle GF (1991). ”The geography and mortality of the 1918 influenza pandemic”. Bulletin of the History of Medicine. 65 (1): 4–21. PMID 2021692.
- Porras-Gallo M, Davis RA, eds. (2014). ”The Spanish Influenza Pandemic of 1918–1919: Perspectives from the Iberian Peninsula and the Americas”. Rochester Studies in Medical History. 30. University of Rochester Press. ISBN 978-1-58046-496-3. Archived from the original on 22 January 2021. Retrieved 9 November 2020 – via Google Books.
- Price-Smith AT (2008). Contagion and Chaos. Cambridge, MA: MIT Press. ISBN 978-0-262-66203-1.
- Qureshi AI (2016). Ebola Virus Disease: From Origin to Outbreak. Academic Press. ISBN 978-0-12-804242-7 – via Google Books.
- Rice GW (2005). Black November; the 1918 Influenza Pandemic in New Zealand (2nd ed.). University of Canterbury Press. ISBN 978-1-877257-35-3.
- Simonsen L, Clarke MJ, Schonberger LB, Arden NH, Cox NJ, Fukuda K (July 1998). ”Pandemic versus epidemic influenza mortality: a pattern of changing age distribution”. The Journal of Infectious Diseases. 178 (1): 53–60. CiteSeerX 10.1.1.327.2581. doi:10.1086/515616. JSTOR 30114117. PMID 9652423.
- Spinney L (2018). Pale rider: the Spanish flu of 1918 and how it changed the world. London: Vintage. ISBN 978-1-78470-240-3. OCLC 1090305029.
- Starko KM (November 2009). ”Salicylates and pandemic influenza mortality, 1918-1919 pharmacology, pathology, and historic evidence”. Clinical Infectious Diseases. 49 (9): 1405–10. doi:10.1086/606060. PMID 19788357. (summary Archived 9 January 2021 at the Wayback Machine by Infectious Diseases Society of America and ScienceDaily, 3 October 2009)
- Starko KM (2010). ”Reply to Noymer et al” (PDF). Clinical Infectious Diseases. 50 (8): 1203–04. doi:10.1086/651473. Archived (PDF) from the original on 17 April 2020. Retrieved 20 April 2018.
- Taubenberger JK, Reid AH, Janczewski TA, Fanning TG (December 2001). ”Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 356 (1416): 1829–39. doi:10.1098/rstb.2001.1020. PMC 1088558. PMID 11779381.
- Taubenberger JK, Reid AH, Lourens RM, Wang R, Jin G, Fanning TG (October 2005). ”Characterization of the 1918 influenza virus polymerase genes”. Nature. 437 (7060): 889–93. Bibcode:2005Natur.437..889T. doi:10.1038/nature04230. PMID 16208372. S2CID 4405787.
- Taubenberger JK, Morens DM (January 2006). ”1918 Influenza: the mother of all pandemics”. Emerging Infectious Diseases. 12 (1): 15–22. doi:10.3201/eid1201.050979. PMC 3291398. PMID 16494711.
- Vana G, Westover KM (June 2008). ”Origin of the 1918 Spanish influenza virus: a comparative genomic analysis”. Molecular Phylogenetics and Evolution. 47 (3): 1100–10. doi:10.1016/j.ympev.2008.02.003. PMID 18353690.
- Vilensky JA, Foley P, Gilman S (August 2007). ”Children and encephalitis lethargica: a historical review”. Pediatric Neurology. 37 (2): 79–84. doi:10.1016/j.pediatrneurol.2007.04.012. PMID 17675021. Archived from the original on 17 April 2020. Retrieved 22 November 2016.
Aiheesta enemmän
- Beiner G (2006). ”Out in the Cold and Back: New-Found Interest in the Great Flu”. Cultural and Social History. 3 (4): 496–505. doi:10.1191/1478003806cs070ra (inactive 31 October 2021). Archived from the original on 24 July 2020. Retrieved 29 May 2020.
- Brown J (2018). Influenza: The Hundred Year Hunt to Cure the Deadliest Disease in History. New York: Atria. ISBN 978-1-5011-8124-5.
- Chandra S, Christensen J, Likhtman S (November 2020). ”Connectivity and seasonality: the 1918 influenza and COVID-19 pandemics in global perspective”. Journal of Global History. 15 (3): 408–20. doi:10.1017/S1740022820000261. S2CID 228988279.
- Collier R (1974). The Plague of the Spanish Lady – The Influenza Pandemic of 1918–19. Atheneum. ISBN 978-0-689-10592-0.
- Davis KC (2018). More Deadly Than War: The Hidden History of the Spanish Flu and the First World War. Henry Holt and Co. ISBN 978-1250145123.
- Dorratoltaj N (29 March 2018). Carrell H (ed.). ”What the 1918 Flu Pandemic Can Teach Today’s Insurers”. AIR Research and Modeling Group. Archived from the original on 7 November 2020. Retrieved 28 May 2019.
- Duncan K (2003). Hunting the 1918 flu: one scientist’s search for a killer virus. University of Toronto Press. ISBN 978-0-8020-8748-5. Archived from the original on 24 July 2020. Retrieved 3 May 2020.
- Humphries MO (2013). The Last Plague: Spanish Influenza and the Politics of Public Health in Canada. University of Toronto Press. ISBN 978-1-4426-1044-6.
- Johnson N (2006). Britain and the 1918–19 Influenza Pandemic: A Dark Epilogue. London and New York: Routledge. ISBN 978-0-415-36560-4.
- Johnson N (2003). ”Measuring a pandemic: Mortality, demography and geography”. Popolazione e Storia. 4 (2): 31–52.
- Johnson N (2003). ”Scottish ’flu – The Scottish mortality experience of the ’Spanish flu'”. Scottish Historical Review. 83 (2): 216–26. doi:10.3366/shr.2004.83.2.216.
- Kolata G (1999). Flu: The Story of the Great Influenza Pandemic of 1918 and the Search for the Virus That Caused It. New York: Farrar, Straus and Giroux. ISBN 978-0-374-15706-7.
- Little J (2007). If I Die Before I Wake: The Flu Epidemic Diary of Fiona Macgregor, Toronto, Ontario, 1918. Dear Canada. Markham, ON: Scholastic Canada. ISBN 978-0-439-98837-7.
- Noymer A, Garenne M (2000). ”The 1918 influenza epidemic’s effects on sex differentials in mortality in the United States”. Population and Development Review. 26 (3): 565–81. doi:10.1111/j.1728-4457.2000.00565.x. PMC 2740912. PMID 19530360.
- Oxford JS, Sefton A, Jackson R, Innes W, Daniels RS, Johnson NP (February 2002). ”World War I may have allowed the emergence of ”Spanish” influenza”. The Lancet. Infectious Diseases. 2 (2): 111–14. doi:10.1016/S1473-3099(02)00185-8. PMID 11901642.
- Oxford JS, Sefton A, Jackson R, Johnson NP, Daniels RS (December 1999). ”Who’s that lady?”. Nature Medicine. 5 (12): 1351–52. doi:10.1038/70913. PMID 10581070. S2CID 5065916.
- Pettit D, Janice B (2008). A Cruel Wind: Pandemic Flu in America, 1918–1920. Murfreesboro, TN: Timberlane Books. ISBN 978-0-9715428-2-2.
- Philips H (2010). ”The re-appearing shadow of 1918: trends in the historiography of the 1918–19 influenza pandemic”. Canadian Bulletin of Medical History. 21 (1): 121–34. doi:10.3138/cbmh.21.1.121. PMID 15202430.
- Phillips H (1 September 2014). ”The Recent Wave of ’Spanish’ Flu Historiography”. Social History of Medicine. 27 (4): 789–808. doi:10.1093/shm/hku066.
- Phillips H, Killingray D, eds. (2003). The Spanish Flu Pandemic of 1918: New Perspectives. London and New York: Routledge.
- Phillips H (November 2020). ”’17,’18,’19: religion and science in three pandemics, 1817, 1918, and 2019”. Journal of Global History. 15 (3): 434–43. doi:10.1017/S1740022820000315. S2CID 228999483.
- Potter CW (October 2001). ”A history of influenza”. Journal of Applied Microbiology. 91 (4): 572–79. doi:10.1046/j.1365-2672.2001.01492.x. PMID 11576290. S2CID 26392163.
- Rice GW, Edwina P (1993). ”Pandemic Influenza in Japan, 1918–1919: Mortality Patterns and Official Responses”. Journal of Japanese Studies. 19 (2): 389–420. doi:10.2307/132645. JSTOR 132645.
- Tumpey TM, García-Sastre A, Mikulasova A, Taubenberger JK, Swayne DE, Palese P, Basler CF (October 2002). ”Existing antivirals are effective against influenza viruses with genes from the 1918 pandemic virus”. Proceedings of the National Academy of Sciences of the United States of America. 99 (21): 13849–54. Bibcode:2002PNAS…9913849T. doi:10.1073/pnas.212519699. PMC 129786. PMID 12368467.
Videot
- We Heard the Bells: The Influenza of 1918 – NIH interviews with Spanish Flu survivors.
- Presentation by Nancy Bristow on the Influenza Pandemic and World War I, November 1, 2019, C-SPAN
- ”Spanish Flu: a warning from history”. Cambridge University. 30 November 2018. Archived from the original on 27 October 2021.
Lähteet ja viitteet
- Yang W, Petkova E, Shaman J (March 2014). ”The 1918 influenza pandemic in New York City: age-specific timing, mortality, and transmission dynamics”. Influenza and Other Respiratory Viruses. National Institutes of Health. 8 (2): 177–88. doi:10.1111/irv.12217. PMC 4082668. PMID 24299150.
- Taubenberger & Morens 2006.
- ”Pandemic Influenza Risk Management WHO Interim Guidance” (PDF). World Health Organization. 2013. p. 25. Archived (PDF) from the original on 21 January 2021. Retrieved 21 August 2021.
- Spreeuwenberg P, Kroneman M, Paget J (December 2018). ”Reassessing the Global Mortality Burden of the 1918 Influenza Pandemic”. American Journal of Epidemiology. Oxford University Press. 187 (12): 2561–67. doi:10.1093/aje/kwy191. PMC 7314216. PMID 30202996.
- Rosenwald MS (7 April 2020). ”History’s deadliest pandemics, from ancient Rome to modern America”. The Washington Post. Archived from the original on 7 April 2020. Retrieved 11 April 2020.
- Mayer J (29 January 2019). ”The Origin Of The Name ’Spanish Flu'”. Science Friday. Retrieved 30 July 2021. Etymology: In ancient times, before epidemiology science, people believed the stars and “heavenly bodies” flowed into us and dictated our lives and health—influenza means ’to influence’ in Italian, and the word stems from the Latin for ’flow in.’ Sickness, like other unexplainable events, was attributed to the influence of the stars… But the name for the infamous 1918 outbreak, the Spanish flu, is actually a misnomer.
- Gagnon, Miller & et al 2013, p. e69586.
- More AF, Loveluck CP, Clifford H, Handley MJ, Korotkikh EV, Kurbatov AV, et al. (September 2020). ”The Impact of a Six-Year Climate Anomaly on the ”Spanish Flu” Pandemic and WWI”. GeoHealth. 4 (9): e2020GH000277. doi:10.1029/2020GH000277. PMC 7513628. PMID 33005839.
- Barry 2004b.
- MacCallum WG (1919). ”Pathology of the pneumonia following influenza”. Journal of the American Medical Association. 72 (10): 720–23. doi:10.1001/jama.1919.02610100028012. Archived from the original on 25 January 2020. Retrieved 16 August 2019.
- Hirsch EF, McKinney M (1919). ”An epidemic of pneumococcus broncho-pneumonia”. Journal of Infectious Diseases. 24 (6): 594–617. doi:10.1093/infdis/24.6.594. JSTOR 30080493. Archived from the original on 24 July 2020. Retrieved 24 May 2020.
- Brundage JF, Shanks GD (December 2007). ”What really happened during the 1918 influenza pandemic? The importance of bacterial secondary infections”. The Journal of Infectious Diseases. 196 (11): 1717–18, author reply 1718–19. doi:10.1086/522355. PMID 18008258.
- Morens DM, Fauci AS (April 2007). ”The 1918 influenza pandemic: insights for the 21st century”. The Journal of Infectious Diseases. 195 (7): 1018–28. doi:10.1086/511989. PMID 17330793.
- ”Influenza Pandemic Plan. The Role of WHO and Guidelines for National and Regional Planning” (PDF). World Health Organization. April 1999. pp. 38, 41. Archived (PDF) from the original on 3 December 2020.
- Michaelis M, Doerr HW, Cinatl J (August 2009). ”Novel swine-origin influenza A virus in humans: another pandemic knocking at the door”. Medical Microbiology and Immunology. 198 (3): 175–83. doi:10.1007/s00430-009-0118-5. PMID 19543913. S2CID 20496301. Archived from the original on 15 May 2021. Retrieved 26 July 2021.
- Mermel LA (June 2009). ”Swine-origin influenza virus in young age groups”. Lancet. 373 (9681): 2108–09. doi:10.1016/S0140-6736(09)61145-4. PMID 19541030. S2CID 27656702. Archived from the original on 27 July 2021. Retrieved 5 April 2021.
- Liang ST, Liang LT, Rosen JM (May 2021). ”COVID-19: a comparison to the 1918 influenza and how we can defeat it”. Postgraduate Medical Journal. 97 (1147): 273–274. doi:10.1136/postgradmedj-2020-139070. PMC 8108277. PMID 33563705. Archived from the original on 10 July 2021. Retrieved 10 July 2021.
- Davis 2013, p. 7: ”In short, although the Spanish flu ’had nothing ”Spanish” about it,’ from a strictly epidemiological perspective, its discursive link to the Iberian nation is beyond dispute. The importance of narrative for a historically informed appreciation of the epidemic stems from the importance of this discursive link and is tied directly to the scientific uncertainty about the etiological agent of the epidemic;”
- ”Biblical Hebrew Words and Meaning”. hebrewversity. 14 March 2018. Retrieved 11 August 2021. in the original Hebrew the people of Israel asked: ”Ma’n Hu?” {?מן הוא} – English for ’what is it?’ and that is the origin of the name ’manna’
- Spinney 2018, p. 65: ”In Freetown, a newspaper suggested that the disease be called manhu until more was known about it. Manhu, a Hebrew word meaning ’what is it?’
- Cole F (1994), Sierra Leone and World War 1, University of London, School of Oriental and African Studies, p. 213, 10731720, retrieved 11 August 2021 – via ProQuest Dissertations Publishing, local interpretations of the crisis had become deeply reminiscent of the increasing disobedience of the Israelites in the wilderness of Sin. It was therefore urged that the epidemic be called ’Man hu,’ (an obvious corruption of ”manna”) meaning ’what is it?’
- Sierra Leone Weekly News, XXXV, 9 July 1918, p. 6 quoted in Mueller JW (1998), p. 8.
- Hammond JA, Rolland W, Shore TH (14 July 1917). ”Purulent Bronchitis”. The Lancet. 190 (4898): 41–46. doi:10.1016/s0140-6736(01)56229-7. ISSN 0140-6736. Alt URL
- Radusin M (September 2012). ”The Spanish flu–Part I: the first wave”. Vojnosanitetski Pregled. 69 (9): 812–817. ISSN 0042-8450. PMID 23050410. the Purple Death … name resulted from the specific skin colour in the most severe cases of the diseased, who by the rule also succumbed to the disease, and is also at the same time the only one which does not link this disease with the Iberian peninsula. This name is actually the most correct.
- McCord CP (8 November 1966). ”The Purple Death. Some things remembered about the influenza epidemic of 1918 at one army camp”. Journal of Occupational Medicine. Industrial Medical Association. 8 (11): 593–598. ISSN 0096-1736. PMID 5334191.
- Getz D (19 September 2017). Purple death : the mysterious Spanish flu of 1918. ISBN 978-1-250-13909-2. OCLC 1006711971.
- Oxford JS, Gill D (2 September 2019). ”A possible European origin of the Spanish influenza and the first attempts to reduce mortality to combat superinfecting bacteria: an opinion from a virologist and a military historian”. Human Vaccines & Immunotherapeutics. 15 (9): 2009–2012. doi:10.1080/21645515.2019.1607711. ISSN 2164-5515. PMC 6773402. PMID 31121112. Two papers were published in The Lancet in 1917 describing an outbreak of disease constituting ’almost a small epidemic’. The first paper was written by physicians at a hospital center in northern France,3 and the second by a team at an army hospital in Aldershot, in southern England. In both earlier instances and in the 1918 pandemic the disease was characterized by a ’dusky’ cyanosis, a rapid progression from quite minor symptoms to death
- Cox J, Gill D, Cox F, Worobey M (1 April 2019). ”Purulent bronchitis in 1917 and pandemic influenza in 1918”. The Lancet Infectious Diseases. 19 (4): 360–361. doi:10.1016/s1473-3099(19)30114-8. ISSN 1473-3099. PMID 30938298. S2CID 91189842.
- Honigsbaum M (23 June 2018). ”Spanish influenza redux: revisiting the mother of all pandemics”. The Lancet. 391 (10139): 2492–2495. doi:10.1016/S0140-6736(18)31360-6. PMID 29976462. S2CID 49709093. Labelled ’purulent bronchitis’ for want of a better term, the disease proved fatal in half the cases and many soldiers also developed cyanosis. 2 years later, British respiratory experts, also writing in The Lancet, but this time in the wake of the pandemic, would decide the disease had been ’fundamentally the same condition’ as ’Spanish’ influenza
- Shanks GD, MacKenzie A, Waller M, Brundage JF (27 November 2011). ”Relationship between ’purulent bronchitis’ in military populations in Europe prior to 1918 and the 1918-1919 influenza pandemic: Precursors of pandemic influenza”. Influenza and Other Respiratory Viruses. 6 (4): 235–239. doi:10.1111/j.1750-2659.2011.00309.x. PMC 5779808. PMID 22118532.
- Storey, C (21 September 2020). ”OLD NEWS: Influenza was an old foe long before 1918”. Arkansas Democrat-Gazette. Opinion. Retrieved 7 August 2021. I came across this seemingly astute analysis in the Dec. 21, 1913, Gazette…. It was a ’Special Cable to the Gazette Through the International News Service.’ Grip Is a Disease Without a Country; All Nations Repudiate Malady: Each Blaming Other Kingdoms, London, Dec. 20. — The grip is a disease without a country, according to a new book just issued which is devoted to the malady. Every country tries to make it out a native of another land…. Eighteenth-century Italian writers say Dr. Hopkirk spoke of ”una influenza di freddo” (influence of cold), and English physicians, mistaking the word influenza for the name of the disease itself, used it. The same term is also used in Germany, where a host of dialect names still prevail, such as lightning catarrh and fog plague.
- ”Grippe is not new”. Los Angeles Herald. 14 January 1899. p. 6 col. 6. Retrieved 7 August 2021 – via California Digital Newspaper Collection. French doctors gave it the name of ”la grippe,” which is now anglicized into ”the grip” … It is known all over the world, and there is a disposition in every nation to shift the odium of it upon some other country. Then the Russians call it the Chinese catarrh, the Germans often call it the Russian pest, the Italians name it the German disease, and the French call it sometimes the Italian fever and sometimes the Spanish catarrh.
- Davis 2013, p. 27: ”On May 21, 1918, El Liberal published a scant article titled ’Can One Live? The Fashionable Illness.’ Likely the first account in Spain of the Spanish flu, it begins: ’For several days, Madrid has been affected by an epidemic, which fortunately is mild; but which, from what it appears, intends to kill doctors from overwork’…. The following day, El Sol and ABC published their first stories about the epidemic: ’What is the Cause? An Epidemic in Madrid’ and ’Benign Epidemic. The Sickbay in Madrid,’ respectively.”
- Alfeirán X (7 December 2015). ”La fiebre de los tres días” [The three-day fever]. La Voz de Galicia (in Spanish). A Coruña. Retrieved 29 July 2021. Las primeras noticias aparecieron en la prensa de Madrid el 21 de mayo de 1918. [The first news appeared in the press of Madrid on 21 May 1918.]
- McClure J (1 May 2009). ”Spanish flu of 1918 no three-day fever. Try 365-day worldwide plague”. York Daily Record. Retrieved 29 July 2021.
- Calleja S (29 July 2021). ”La fiebre de los tres días” [The three-day fever]. Diario de León (in Spanish). León, Spain. Retrieved 21 October 2018.
- ”Impacto de la gripe de 1918 en España”. Comité Asesor de Vacunas de la Asociación Española de Pediatría [Vaccine Advisory Committee of the Spanish Association of Pediatrics] (in Spanish). Retrieved 29 July 2021. En España, se le llamó al principio ’la fiebre de los tres días’, atendiendo a la creencia, como en otros países, de que la gripe era una enfermedad leve. Las primeras noticias sobre la gripe, llamando la atención sobre que algo distinto estaba ocurriendo, aparecieron en la prensa a finales de mayo. Por ej. en el diario ABC el 22 de mayo mediante una escueta nota en la página 24: ’Los médicos han comprobado, en Madrid, la existencia de una epidemia de índole gripal, muy propagada, pero, por fortuna, de carácter leve’. [In Spain, it was initially called ’the three-day fever’, based on the belief, as in other countries, that the flu was a mild illness. The first reports about the flu, drawing attention to the fact that something different was happening, appeared in the press at the end of May. For example, in the newspaper ABC on 22 May, through a brief note on page 24: ”The doctors have verified, in Madrid, the existence of an epidemic of a influenza nature, very widespread, but, fortunately, of a mild nature.’]
- Killingray D, Phillips H (2 September 2003). The Spanish Influenza Pandemic of 1918-1919: New Perspectives. Routledge. ISBN 978-1-134-56640-2. In the popular mind calamities often need to have their origin and cause identified and other countries or peoples credited with blame. This xenophobic response has been common in Europe, that impulse to blame others or the silent places of the Asian heartlands for the source of disease.
- Hoppe T (November 2018). ””Spanish Flu”: When Infectious Disease Names Blur Origins and Stigmatize Those Infected”. American Journal of Public Health. 108 (11): 1462–1464. doi:10.2105/AJPH.2018.304645. PMC 6187801. PMID 30252513.
- Bax D (2020). ”Pandemie – Welt im Fieber” [Pandemic – World in Fever]. www.freitag.de (in German) (13). Retrieved 31 July 2021. In Großbritannien wurde die Krankheit dagegen als ’Flandrische Grippe’ bezeichnet, weil sich viele —en in den Schützengräben von Flandern ansteckten. [In Britain, on the other hand, the disease was called the ’Flanders flu’ because many soldiers became infected in the trenches of Flanders.]
- Reynolds JR (1866). A System of Medicine. 1. Macmillan. pp. 30–31. One is, that every epidemic owns one unknown source, whence it spreads; each nation, in turn, attributing to its neighbour from whom it derived the disease, the unenviable honour of originating it. Thus the Italians have termed it the German disease; the Germans, the Russian pest; the Russians, the Chinese Catarrh;
- Harrison MA, Claiborne JH Jr, eds. (January 1890). ”Editorials: La Grippe”. Gaillard’s Medical Journal. 50: 217. The outbreak immediately preceding the present one was in 1879, and has been well described by Da Costa. Like every other disease about whose pathology we know very little, this malady has not a scientifically correct name, but at different times and in different countries has received names which are almost purely local; thus the Russians have called it the Chinese catarrh, because it has often invaded Russia from China. The Germans call it the Russian pest, while the Italians in turn call it the German disease.
- Davis KC (2018). More deadly than war : the hidden history of the Spanish flu and the First World War (First ed.). New York. ISBN 978-1-250-14512-3. OCLC 1034984776. The Russians called it the Chinese flu. In Japan, it was wrestler’s fever. In South Africa, it was known as either the white man’s sickness or kaffersiekte blacks’ disease. Soldiers fighting in the Great War called it the three-day fever—a highly inaccurate description—and when it first struck in the spring of 1918, German soldiers called it Flanders fever, after one of the war’s most notorious and deadly battlefields
- Porras-Gallo & Davis 2014, p. 51.
- Galvin 2007.
- Own Correspondent (2 June 1918). ”The Spanish Epidemic”. The Times. London. Retrieved 29 July 2021. The unknown disease which appeared in Madrid a fortnight ago spread with remarkable rapidity…. It is reported that there are well over 100,000 victims in Madrid alone…. Although the disease is clearly of a gripal character…
- ”The Spanish Influenza; A Sufferer’s Symptoms”. The Times. London. 25 June 1918. Retrieved 29 July 2021.
- ”Public Notice; ’Spanish Influenza’ Epidemic”. The Times. London. 28 June 1918. Retrieved 11 August 2021.
- Arnold C (13 September 2018). ”Eat More Onions!”. Lapham’s Quarterly. Retrieved 12 August 2021. daily newspapers carried an increasing number of advertisements for influenza-related remedies as drug companies played on the anxieties of readers and reaped the benefits. From the Times of London to the Washington Post, page after page was filled with dozens of advertisements for preventive measures and over-the-counter remedies. ’Influenza!’ proclaimed an advert extolling the virtues of Formamint lozenges.
- Daniels R (29 April 2018) [1998 Mill Hill Essays]. ”In Search of an Enigma: The ”Spanish Lady””. NIMR History. Archived from the original on 29 April 2018. Retrieved 9 August 2021 – via web.archive.org. In turn, when Russia reported on the situation in Moscow, Pravda printed ”Ispanka (The Spanish Lady) is in town” and the name has stuck.
- Trilla A, Trilla G, Daer C (September 2008). ”The 1918 ”Spanish flu” in Spain”. Clinical Infectious Diseases. 47 (5): 668–73. doi:10.1086/590567. PMID 18652556.
- ”Spanish flu facts”. Channel 4 News. Archived from the original on 27 January 2010.
- Anderson S (29 August 2006). ”Analysis of Spanish flu cases in 1918–1920 suggests transfusions might help in bird flu pandemic”. American College of Physicians. Archived from the original on 25 November 2011. Retrieved 2 October 2011.
- Barry 2004, p. 171.
- Spinney L (29 September 2018). ”The centenary of the 20th century’s worst catastrophe”. The Economist. Science & technology. ISSN 0013-0613. Retrieved 3 August 2021. On June 29th 1918 Martín Salazar, Spain’s inspector-general of health, stood up in front of the Royal Academy of Medicine in Madrid. He declared, not without embarrassment, that the disease which was ravaging his country was to be found nowhere else in Europe. In fact, that was not true. The illness in question, influenza, had been sowing misery in France and Britain for weeks, and in America for longer, but Salazar did not know this because the governments of those countries, a group then at war with Germany and its allies, had made strenuous efforts to suppress such potentially morale-damaging news.
- ”Madrid Letter”. Journal of the American Medical Association. 71 (19): 1594. 9 November 1918.
- Radusin M (September 2012). ”The Spanish flu – Part I: the first wave”. Vojnosanitetski Pregled. 69 (9): 812–817. ISSN 0042-8450. PMID 23050410. ’The Serbian Newspaper’ wrote in the period October 23 / November 5, 1918 at the peak of the second, deadliest wave of the pandemic: ’Various countries have been assigning the origin of this imposing guest to each other for quite some time, and at one point in time they agreed to assign its origin to the kind and neutral Spain, which had been fighting off this honour just as much as German submarines, so imposing in their search for hospitality off Spanish shores.’
- Vázquez-Espinosa E, Laganà C, Vázquez F (October 2020). ”The Spanish flu and the fiction literature”. Revista Espanola de Quimioterapia. 33 (5): 296–312. doi:10.37201/req/049.2020. PMC 7528412. PMID 32633114. French journalists had, initially, called it the ’American flu’; but the fact that the American soldiers were his allies in the warlike conflict advised not to assign such a link to them…. Another most popular name in Madrid, was the ’Soldado de Nápoles’ (Naples soldier), a popular song in the zarzuela (popular musical genre or ’género chico’ in Spain) called La canción del olvido (The forgotten song) due both, were ’highly contagious’. Today, there are many authors who avoid such a name (the Spanish flu) and they aptly refer to it as the ’1918- 1819[sic] influenza pandemic’
- Müller S (2020). ”Spanische Grippe” [The Spanish Flu]. www.fes.de (in German). Bonn: Friedrich Ebert Foundation. Retrieved 31 July 2021. In Europa wurde die Spanischer Grippe auch als ’Blitzkatarrh’, als ’Flandern-Fieber’, ’flandische Grippe’, bei Engländern und Amerikanern als ’three-day’- oder ’knock-me-down’-Fieber, und in Frankreich als ’la grippe’, als ’bronchite purulente’ (eitrige Bronchitis) oder beim französische Militärärzte als ’Krankheit 11’ (maladie onze) bezeichnet. Die Benennung von Krankheiten und insbesondere Seuchen nach ihrem vermuteten Ursprungsort ist nichts Ungewöhnliches. Es ist der Versuch, einem Geschehen auf die Spur zu kommen. Zugleich werden auf diese Weise Krankheiten als etwas Äußerliches gekennzeichnet, als etwas Fremdes, das eingedrungen ist oder eingeschleppt wurde. [In Europe, the Spanish flu was also referred to as ’Blitzkatarrh’, as ’Flanders fever’, ’Flanders flu’, in English and Americans as ’three-day’ or ’knock-me-down’ fever, and in France as ’la flu’, as ’bronchite purulente’ purulent bronchitis) or by French military doctors as ’disease 11’ (maladie onze). The naming of diseases and especially epidemics according to their presumed place of origin is nothing unusual. It is an attempt to track down what is happening. At the same time, in this way, diseases are marked as something external, as something foreign that has invaded or been introduced.]
- Cotter C (23 April 2020). ”From the ’Spanish Flu’ to COVID-19: lessons from the 1918 pandemic and First World War”. Humanitarian Law & Policy. International Committee of the Red Cross. Retrieved 7 August 2021. Many other nicknames were given to the pandemic, many based on nationality or race: ’Spanish Lady’, ’French Flu’, ’Naples Soldier’, ’Purple Death’, ’War Plague’, ’Flanders Grippe’, ’Kirghiz Disease’, ’Black Man’s Disease’, ’Hun Flu’, ’German Plague’, ’Bolshevik Disease’ or even the ’Turco-Germanic bacterium criminal entreprise’. These discriminatory epithets reflect the many rumors and theories that quickly spread about the origins of the pathology.
- Spinney 2018, p. 58.
- Davis 2013, pp. 103–36.
- Landgrebe P (29 December 2018). ”100 Years After: The Name of Death”. History Campus. Archived from the original on 16 August 2020. Retrieved 16 August 2020.
- Rodríguez LP, Palomba AL (5 March 2019). ”How is the adjective ’Spanish’ used in other languages?”. El País (English ed.). Madrid. Retrieved 1 August 2021. The use of ’Spanish’ can often have negative connotations, with the adjective often unfairly used to describe unwelcome events and problems. The most obvious example is the so-called ’Spanish flu,’ a reference to the 1918 influenza pandemic…
- O’Reilly T (13 May 2020). ”How the Spanish Flu wasn’t Spanish at all”. CBC Radio. Under the Influence. Retrieved 1 August 2021. Medical professionals and officials in Spain protested. They said the Spanish people were being falsely stigmatized…. If you’ve ever wondered about the staying power of a brand, the ’Spanish Flu’ is a case in point. A full 100 years later, the ’Spanish Flu’ is still referenced — and still remains a source of irritation in Spain.
- Takon L (7 April 2020). ”Fighting words: how war metaphors can trigger racism”. The Lighthouse. Macquarie University. Retrieved 8 August 2021. the names given to this disease in different parts of the world reflected prevailing concerns about certain ethnic groups and ideologies. The disease was called the ’Singapore fever’ in Penang and the Bolshevik disease in Poland.
- Phillips, H. (1990). Black October: The Impact of the Spanish Influenza Epidemic of 1918 on South Africa. Government Printer, South Africa. ISBN 978-0-7970-1580-7. In one area where Blacks were the first victims, the accusatory term ’Kaffersiekte’ was coined; in another district, where the position was reversed, Blacks returned the compliment with, White man’s sickness’
- 余録:「春のさきぶれ」といえば何か聞こえが良いが… [Speaking of ”spring sekibu”, something sounds good…]. =毎日新聞 [Mainichi Shimbun morning newspaper] (in Japanese). 15 May 2020. Retrieved 8 August 2021. 翌月の東京の夏場所は高熱などによる全休力士が相次いだ。世間はこれを「相撲風邪」「力士風邪」と呼んだが、実はこの謎の感染症こそが同年初めから米国で流行の始まった「スペイン風邪」とみられている。 [The following month, a number of sumo wrestlers were absent from the summer tournament in Tokyo due to high fevers. People called it the ”sumo flu” or ”wrestler flu,” but in fact, this mysterious infection is believed to be the Spanish flu, which began spreading in the United States early that year.]
- Suzuki K (6 April 2020). ”The Spanish Flu in Japan and Its Parallels to COVID-19”. Unseen Japan. Retrieved 8 August 2021. The 1918 Influenza epidemic began in Japan in late August 1918 and became a national epidemic in November. Experts believe it entered Japan through a group of sumo wrestlers returning from Taiwan (a colony of Japan at the time). Because of this, for a while, people in Japan called it the ’sumo flu.’
- ”How the Spanish flu of 1918-20 ravaged Japan”. Japan Today. Retrieved 8 August 2021. The first patients in Japan, reported Shukan Gendai (May 2–9), began showing symptoms around April 1918. Initially the disease was referred to as the ”Sumo Kaze” (sumo cold) because a contingent of sumo wrestlers contracted it while on a tour of Taiwan. Three well known grapplers, Masagoishi, Choshunada and Wakagiyama, died before they could return from Taiwan. As the contagion spread, the summer sumo tournament, which would have been held on the grounds of Yasukuni shrine, was cancelled.
- Dionne & Turkmen 2020, pp. 213–230.
- ”WHO issues best practices for naming new human infectious diseases”. www.who.int. 8 May 2015. Retrieved 4 August 2021.
- ”Pandemic influenza: an evolving challenge”. World Health Organization. 22 May 2018. Archived from the original on 20 March 2020. Retrieved 20 March 2020.
- ”Influenza pandemic of 1918–19”. Encyclopaedia Britannica. 4 March 2020. Archived from the original on 20 March 2020. Retrieved 20 March 2020.
- Chodosh S (18 March 2020). ”What the 1918 flu pandemic can teach us about COVID-19, in four charts”. PopSci. Archived from the original on 24 December 2020. Retrieved 20 March 2020.
- Mueller JW (1998). What’s in a name. Spanish Influenza in sub-Saharan Africa and what local names say about the perception of this pandemic. Spanish ’Flu 1918–1998: Reflections on the Influenza Pandemic of 1918 after 80 Years. Cape Town, South Africa. Retrieved 13 August 2021.
- Phillips H (8 October 2014). ”Influenza Pandemic (Africa)”. encyclopedia.1914-1918-online.net. International Encyclopedia of the First World War (WW1). Retrieved 13 August 2021.
- Great Britain Ministry of Health (1920). Report on the Pandemic of Influenza, 1918–19. H.M. Stationery Office. There was a high west wind at the time and this was thought to be the carrying agent so that the affliction was called ’the disease of the wind.’
- Afkhami AA (5 February 2019). A Modern Contagion: Imperialism and Public Health in Iran’s Age of Cholera. JHU Press. ISBN 978-1-4214-2722-5. In Tehran, the disease was called the ”illness of the wind” (nakhushi-yi bad) due to its initial occurrence during a strong westerly wind burst and its rapid spread.
- Archives of Internal Medicine. 24. Chicago: American Medical Association. 1919. As this included the period of the great influenza epidemic when Type IV infections were usually prevalent, it will be seen that the proportion of fixed types among carriers and among cases is not far from the same.
- United States Surgeon-General’s Office (1920). Report of the Surgeon-General of the Army to the Secretary of War for the Fiscal Year Ending June 30, 1919. War Department Annual Reports. I. U.S. Government Printing Office. On September 13, 1918, the first cases of the great influenza epidemic were admitted, and during the next 10 weeks over 4,100 patients were admitted.
- Spinney 2018, p. 64: ”In … Rhodesia (Zimbabwe) … officials labelled the new affliction ’influenza (vera)’, adding the Latin word vera, meaning ’true’ … German doctors … called it ’pseudo-influenza'”
- Cross A (29 March 2020). ”A look at art and music created in times of pandemic”. Global News (Canada). Retrieved 9 August 2021. Another rhyme with deadly origins appeared during a worldwide influenza pandemic in 1889-1890. Certain the disease could be stopped by sealing up the home from the poisoned air outside, this safety tip emerged in schools: There was a little girl, and she had a little bird; And she called it by the pretty name of Enza; But one day it flew away, but it didn’t go to stay; For when she raised the window, in-flu-Enza
- University Libraries. ”WWI Exhibit: Influenza Pandemic”. content.lib.washington.edu. University of Washington. Retrieved 9 August 2021.
- Hakim J (September 2002). War, Peace, and All that Jazz. Oxford University Press. ISBN 978-0-19-515335-4. In flew Enza—say it fast and it becomes ’influenza.’ It was a catchy little rhyme, and boys and girls skipped rope to it.
- Honigsbaum M (18 October 2016). Living with Enza: The Forgotten Story of Britain and the Great Flu Pandemic of 1918. Springer. ISBN 978-0-230-23921-0. A popular playground skipping rhyme caught the ease with which this ’new disease’ was transmitted: I had a little bird; Its name was Enza; I opened the window; And in-flu-enza. As influenza spread from Glasgow to Aberdeen, and from Liverpool to London’s Mile End, it was not long before playgrounds throughout the country echoed to the chant.
- March PC (4 September 1932). ”General March’s Narrative: Glimpses of Woodrow Wilson”. The New York Times. p. XX3, Special Features section.
- ”The 1918 Flu Pandemic Killed Hundreds of Thousands of Americans. The White House Never Said a Word About It”. Time. Retrieved 10 August 2021. Crosby’s America’s Forgotten Pandemic: The Influenza of 1918, Wilson asked Army Chief of Staff General Peyton March in October 1918 if he had heard of the popular jump rope rhyme parodying the virus, and recited part of it.
- Spinney 2018, p. 36.
- ”1918 Flu (Spanish flu epidemic)”. Avian Bird Flu. Archived from the original on 21 May 2008.
- Sheidlower N (17 March 2020). ”How NYC Survived the 1918 Spanish Flu Pandemic”. Untapped New York. Archived from the original on 29 September 2020. Retrieved 8 October 2020.
- Billings 1997.
- ”The Memoirs of Herbert Hoover: Years of Adventure, 1874–1920. (New York: Macmillan Company. 1951. pp. xi, 496.) and Herbert Hoover and the Russian Prisoners of World War I: A Study in Diplomacy and Relief, 1918–1919. By Edward F. Willis. (Stanford: Stanford University Press. 1951. pp. viii, 67.)”. The American Historical Review: 12. 2011. doi:10.1086/ahr/57.3.709. ISSN 1937-5239.
- Spinney 2018, p. 37.
- ”Queer Epidemic Sweeps North China; Banks and Silk Stores in Peking Closed – Another Loan Sought from Japan”. The New York Times. 1 June 1918. ISSN 0362-4331. Archived from the original on 24 June 2020. Retrieved 22 June 2020.
- Patterson & Pyle 1991.
- ”Mortality Statistics 1918: Nineteenth Annual Report” (PDF). United States Census Bureau. 1920. p. 28. Archived (PDF) from the original on 11 July 2020. Retrieved 29 May 2020.
- Erkoreka A (March 2010). ”The Spanish influenza pandemic in occidental Europe (1918–1920) and victim age”. Influenza and Other Respiratory Viruses. 4 (2): 81–89. doi:10.1111/j.1750-2659.2009.00125.x. PMC 5779284. PMID 20167048.
- Spinney 2018, p. 38.
- Byerly CR (April 2010). ”The U.S. military and the influenza pandemic of 1918–1919”. Public Health Reports. 125 (Suppl 3): 82–91. doi:10.1177/00333549101250S311. PMC 2862337. PMID 20568570.
- Spinney 2018, p. 39.
- ”Atatürk işgalcilerden önce İspanyol Gribini yenmişti”. www.sozcu.com.tr (in Turkish). Archived from the original on 8 November 2020. Retrieved 2 November 2020.
- Spinney 2018, p. 40.
- Barry JM (2005). The Great Influenza. United States: Penguin Books. p. 270. ISBN 0-670-89473-7. On September 15, New York City’s first influenza death occurred.
- Flynn M (12 March 2020). ”What happens if parades aren’t canceled during pandemics? Philadelphia found out in 1918, with disastrous results”. The Washington Post. Archived from the original on 4 July 2020. Retrieved 9 July 2020.
- Spinney 2018, p. 41.
- Spinney 2018, p. 42.
- Kenner R (18 January 2010). ”Influenza 1918”. American Experience. Season 10. Episode 5. PBS. WGBH. Archived from the original on 27 May 2020. Transcript. Retrieved 27 May 2020.
- ”Epidemic Influenza”. p. 93. Archived from the original on 4 July 2020. Retrieved 4 July 2020.
- Radusin M (October 2012). ”The Spanish flu – part II: the second and third wave”. Vojnosanitetski Pregled. 69 (10): 917–27. PMID 23155616. Archived from the original on 9 July 2020. Retrieved 23 April 2020.
- ”1918 Pandemic Influenza: Three Waves”. Centers for Disease Control and Prevention. 11 May 2018. Archived from the original on 23 April 2020. Retrieved 23 April 2020.
- Najera RF (2 January 2019). ”Influenza in 1919 and 100 Years Later”. College of Physicians of Philadelphia. Archived from the original on 24 July 2020. Retrieved 23 April 2020.
- ”Here are Exact Facts About the Influenza and Its Toll in City, State, Nation, world”. Los Angeles Times. 9 February 1919. Archived from the original on 24 July 2020. Retrieved 10 May 2020.
- Vaughan WT (July 1921). ”Influenza: An Epidemiologic Study”. American Journal of Hygiene. ISBN 978-0-598-84038-7. Archived from the original on 24 July 2020. Retrieved 11 May 2020.
- ”Mortality Statistics 1919: Twentieth Annual Report” (PDF). United States Census Bureau. 1921. p. 30. Archived (PDF) from the original on 12 July 2020. Retrieved 11 May 2020.
- Ansart S, Pelat C, Boelle PY, Carrat F, Flahault A, Valleron AJ (May 2009). ”Mortality burden of the 1918–1919 influenza pandemic in Europe”. Influenza and Other Respiratory Viruses. 3 (3): 99–106. doi:10.1111/j.1750-2659.2009.00080.x. PMC 4634693. PMID 19453486.
- ”How the 1918 flu pandemic rolled on for years: a snapshot from 1920”. The Guardian. Archived from the original on 6 April 2020. Retrieved 30 April 2020.
- Vaughan WT (July 1921). ”Influenza: An Epidemologic Study”. The American Journal of Hygiene. p. 91. Archived from the original on 6 October 2020. Retrieved 13 August 2020.
- Spinney 2018, p. 43.
- [1]
- Crosby 2003.
- Worobey M, Cox J, Gill D (2019). ”The origins of the great pandemic”. Evolution, Medicine, and Public Health. 2019 (1): 18–25. doi:10.1093/emph/eoz001. PMC 6381288. PMID 30805187. S2CID 67863937.
- ”Grippe : un virus plurimillénaire et ravageur découvert seulement en 1933” [Influenza: a multi-millennial and pest virus discovered only in 1933]. France Culture (in French). 16 January 2019. Retrieved 29 July 2021.
- Valentine V (20 February 2006). ”Origins of the 1918 Pandemic: The Case for France”. National Public Radio. Archived from the original on 30 April 2009. Retrieved 13 April 2020.
- Oxford JS (December 2001). ”The so-called Great Spanish Influenza Pandemic of 1918 may have originated in France in 1916”. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. Royal Society. 356 (1416): 1857–59. doi:10.1098/rstb.2001.1012. PMC 1088561. PMID 11779384.
- Connor S (8 January 2000). ”Flu epidemic traced to Great War transit camp”. The Guardian. UK. Archived from the original on 12 May 2009. Retrieved 9 May 2009.
- Oxford JS, Lambkin R, Sefton A, Daniels R, Elliot A, Brown R, Gill D (January 2005). ”A hypothesis: the conjunction of soldiers, gas, pigs, ducks, geese and horses in northern France during the Great War provided the conditions for the emergence of the ”Spanish” influenza pandemic of 1918–1919″ (PDF). Vaccine. 23 (7): 940–45. doi:10.1016/j.vaccine.2004.06.035. PMID 15603896. Archived from the original (PDF) on 12 March 2020. Retrieved 12 March 2020.
- Shanks GD (January 2016). ”No evidence of 1918 influenza pandemic origin in Chinese laborers/soldiers in France”. Journal of the Chinese Medical Association. 79 (1): 46–48. doi:10.1016/j.jcma.2015.08.009. PMID 26542935.
- Price-Smith 2008.
- Ansart S, Pelat C, Boelle PY, Carrat F, Flahault A, Valleron AJ (May 2009). ”Mortality burden of the 1918–1919 influenza pandemic in Europe”. Influenza and Other Respiratory Viruses. Wiley. 3 (3): 99–106. doi:10.1111/j.1750-2659.2009.00080.x. PMC 4634693. PMID 19453486.
- Hannoun C (1993). ”La Grippe”. Documents de la Conférence de l’Institut Pasteur. La Grippe Espagnole de 1918. Ed Techniques Encyclopédie Médico-Chirurgicale (EMC), Maladies infectieuses. 8-069-A-10.
- Humphries 2014.
- Vergano D (24 January 2014). ”1918 Flu Pandemic That Killed 50 Million Originated in China, Historians Say”. National Geographic. Archived from the original on 26 January 2014. Retrieved 4 November 2016.
- Spinney 2018, p. 143.
- Killingray D, Phillips H (2003). The Spanish Influenza Pandemic of 1918–1919: New Perspectives. Routledge. ISBN 978-1-134-56640-2. Archived from the original on 9 May 2021. Retrieved 9 November 2020.
- Langford C (2005). ”Did the 1918-19 Influenza Pandemic Originate in China?”. Population and Development Review. 31 (3): 473–505. doi:10.1111/j.1728-4457.2005.00080.x. JSTOR 3401475.
- Cheng KF, Leung PC (July 2007). ”What happened in China during the 1918 influenza pandemic?”. International Journal of Infectious Diseases. 11 (4): 360–64. doi:10.1016/j.ijid.2006.07.009. PMID 17379558.
- Vergano D. ”1918 Flu Pandemic That Killed 50 Million Originated in China, Historians Say”. National Geographic. Archived from the original on 3 March 2020. Retrieved 5 March 2020.
- Saunders-Hastings PR, Krewski D (December 2016). ”Reviewing the History of Pandemic Influenza: Understanding Patterns of Emergence and Transmission”. Pathogens. 5 (4): 66. doi:10.3390/pathogens5040066. PMC 5198166. PMID 27929449.
- Mills CE, Robins JM, Lipsitch M (December 2004). ”Transmissibility of 1918 pandemic influenza”. Nature. 432 (7019): 904–06. Bibcode:2004Natur.432..904M. doi:10.1038/nature03063. PMC 7095078. PMID 15602562. Archived from the original on 25 June 2020. Retrieved 24 June 2020.
- Qureshi 2016, p. 42.
- Ewald 1994.
- ”The Spanish Flu pandemic of 1918”. The History Press. Archived from the original on 13 January 2021. Retrieved 20 February 2021.
- Illing S (20 March 2020). ”The most important lesson of the 1918 influenza pandemic: Tell the damn truth”. Vox. Archived from the original on 25 March 2020. John M. Barry : The government lied. They lied about everything. We were at war and they lied because they didn’t want to upend the war effort. You had public health leaders telling people this was just the ordinary flu by another name. They simply didn’t tell the people the truth about what was happening.
- Gladwell 1997, p. 55.
- Gladwell 1997, p. 63.
- Fogarty International Center. ”Summer Flu Outbreak of 1918 May Have Provided Partial Protection Against Lethal Fall Pandemic”. Fic.nih.gov. Archived from the original on 27 July 2011. Retrieved 19 May 2012.
- Gladwell 1997, p. 56.
- Spinney 2018, pp. 45–47.
- Knobler 2005.
- Morris DE, Cleary DW, Clarke SC (2017). ”Secondary Bacterial Infections Associated with Influenza Pandemics”. Frontiers in Microbiology. 8: 1041. doi:10.3389/fmicb.2017.01041. PMC 5481322. PMID 28690590.
- ”Bacterial Pneumonia Caused Most Deaths in 1918 Influenza Pandemic”. National Institutes of Health. 23 September 2015. Archived from the original on 22 April 2016. Retrieved 17 April 2016.
- Taubenberger et al. 2001, pp. 1829–39.
- Morens DM, Taubenberger JK, Fauci AS (October 2008). ”Predominant role of bacterial pneumonia as a cause of death in pandemic influenza: implications for pandemic influenza preparedness”. The Journal of Infectious Diseases. 198 (7): 962–70. doi:10.1086/591708. PMC 2599911. PMID 18710327.
- Barry 2004.
- Spinney 2018, p. 61.
- ”Washington State Board of Health pessimistic about influenza pandemic in a report to the governor on January 1, 1919”. www.historylink.org. Archived from the original on 21 July 2020. Retrieved 21 July 2020.
- Spinney 2018, p. 62.
- Foxman EF, Storer JA, Fitzgerald ME, Wasik BR, Hou L, Zhao H, et al. (January 2015). ”Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells”. Proceedings of the National Academy of Sciences of the United States of America. 112 (3): 827–32. Bibcode:2015PNAS..112..827F. doi:10.1073/pnas.1411030112. PMC 4311828. PMID 25561542.
- Lowen AC, Steel J (July 2014). ”Roles of humidity and temperature in shaping influenza seasonality”. Journal of Virology. 88 (14): 7692–95. doi:10.1128/JVI.03544-13. PMC 4097773. PMID 24789791.
- Brown JD, Goekjian G, Poulson R, Valeika S, Stallknecht DE (April 2009). ”Avian influenza virus in water: infectivity is dependent on pH, salinity and temperature”. Veterinary Microbiology. 136 (1–2): 20–26. doi:10.1016/j.vetmic.2008.10.027. PMID 19081209.
- Foxman EF, Storer JA, Vanaja K, Levchenko A, Iwasaki A (July 2016). ”Two interferon-independent double-stranded RNA-induced host defense strategies suppress the common cold virus at warm temperature”. Proceedings of the National Academy of Sciences of the United States of America. 113 (30 Suppl 3): 8496–501. doi:10.1073/pnas.1601942113. PMC 4968739. PMID 27402752.
- Klugman KP, Chien YW, Madhi SA (August 2009). ”Pneumococcal pneumonia and influenza: a deadly combination”. Vaccine. 27 (s3): C9–C14. doi:10.1016/j.vaccine.2009.06.007. PMID 19683658.
- Bengtsson D, Safi K, Avril A, Fiedler W, Wikelski M, Gunnarsson G, et al. (February 2016). ”Does influenza A virus infection affect movement behaviour during stopover in its wild reservoir host?”. Royal Society Open Science. 3 (2): 150633. Bibcode:2016RSOS….350633B. doi:10.1098/rsos.150633. PMC 4785985. PMID 26998334.
- Tolf C, Bengtsson D, Rodrigues D, Latorre-Margalef N, Wille M, Figueiredo ME, et al. (2012). ”Birds and viruses at a crossroad – surveillance of influenza A virus in Portuguese waterfowl”. PLOS ONE. 7 (11): e49002. Bibcode:2012PLoSO…749002T. doi:10.1371/journal.pone.0049002. PMC 3492218. PMID 23145046.
- Tucker MA, Böhning-Gaese K, Fagan WF, Fryxell JM, Van Moorter B, Alberts SC, et al. (January 2018). ”Moving in the Anthropocene: Global reductions in terrestrial mammalian movements”. Science. 359 (6374): 466–69. Bibcode:2018Sci…359..466T. doi:10.1126/science.aam9712. PMID 29371471.
- Blakemore E (3 October 2020). ”Catastrophic effect of 1918 flu may have been aided by peculiar influx of cold air into Europe during WWI”. The Washington Post. Archived from the original on 11 December 2020. Retrieved 29 November 2020.
- Kent L (28 September 2020). ”How environmental conditions like cold and wet weather can affect pandemics, and what that means for COVID-19”. CNN. Archived from the original on 2 December 2020. Retrieved 29 November 2020.
- Powell A (5 October 2020). ”Six-year deluge linked to Spanish flu, World War I deaths”. Harvard Gazette. Archived from the original on 27 November 2020. Retrieved 29 November 2020.
- ”Gauze Mask to Halt Spread of Plague”. The Washington Times. 27 September 1918. p. 3. Archived from the original on 8 October 2020. Retrieved 5 October 2020.
- Hauck G, Gellis K (22 November 2020). ”We’re celebrating Thanksgiving amid a pandemic. Here’s how we did it in 1918 – and what happened next”. USA Today. Archived from the original on 21 November 2020.
- Spinney 2018, pp. 83–84.
- Spinney 2018, pp. 87–88.
- Spinney 2018, p. 91.
- ”How NYC beat deadly 1918 flu without vaccines, lockdowns, mask mandates or school closures”. silive. 20 September 2021. Retrieved 23 September 2021.
- Bootsma MC, Ferguson NM (May 2007). ”The effect of public health measures on the 1918 influenza pandemic in U.S. cities”. Proceedings of the National Academy of Sciences of the United States of America. 104 (18): 7588–93. doi:10.1073/pnas.0611071104. PMC 1849868. PMID 17416677. S2CID 11280273.
- Spinney 2018, pp. 109–10.
- Spinney 2018, pp. 111–12.
- Spinney 2018, p. 92.
- Spinney 2018, p. 69.
- Spinney 2018, p. 70.
- Taubenberger & Morens 2006, p. 19.
- Taubenberger & Morens 2006, p. 17.
- ”Ten things you need to know about pandemic influenza (update of 14 October 2005)” (PDF). Relevé Épidémiologique Hebdomadaire. 80 (49–50): 428–31. December 2005. PMID 16372665. Archived (PDF) from the original on 27 July 2021. Retrieved 11 March 2020.
- Jilani TN, Jamil RT, Siddiqui AH (14 December 2019). ”H1N1 Influenza (Swine Flu)”. StatPearls. Treasure Island, FL: StatPearls. PMID 30020613. Archived from the original on 12 March 2020. Retrieved 11 March 2020 – via NCBI.
- Johnson & Mueller 2002.
- Chandra S, Christensen J (July 2019). ”Re: ”Reassessing the Global Mortality Burden of the 1918 Influenza Pandemic””. American Journal of Epidemiology. 188 (7): 1404–06. doi:10.1093/aje/kwz044. PMID 30824934. and response Spreeuwenberg P, Kroneman M, Paget J (July 2019). ”The Authors Reply” (PDF). American Journal of Epidemiology. 188 (7): 1405–06. doi:10.1093/aje/kwz041. PMID 30824908. Archived from the original (PDF) on 12 March 2020. Retrieved 12 March 2020.
- ”Historical Estimates of World Population”. Archived from the original on 9 July 2012. Retrieved 29 March 2013.
- Mayor S (October 2000). ”Flu experts warn of need for pandemic plans”. BMJ. 321 (7265): 852. doi:10.1136/bmj.321.7265.852. PMC 1118673. PMID 11021855.
- Chandra, Kuljanin & Wray 2012.
- David Arnold, ”Dearth and the Modern Empire: The 1918–19 Influenza Epidemic in India,” Transactions of the Royal Historical Society 29 (2019): 181–200.
- Sreevatsan A (12 March 2020). ”Why 1918 matters in India’s corona war”. mint. Archived from the original on 18 April 2020. Retrieved 7 June 2020.
- ”What the history of pandemics tells us about coronavirus”. Hindustan Times. 15 April 2020. Archived from the original on 21 April 2020. Retrieved 7 June 2020.
- Murray CJ, Lopez AD, Chin B, Feehan D, Hill KH (December 2006). ”Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–20 pandemic: a quantitative analysis”. Lancet. Elsevier. 368 (9554): 2211–18. doi:10.1016/S0140-6736(06)69895-4. PMID 17189032. S2CID 22787011. Archived from the original on 27 September 2020. Retrieved 30 September 2020.
- ”Historiallisia Papereita 1”. historiallinenyhdistys.fi. ISSN 1456-8055. Archived from the original on 6 February 2020. Retrieved 24 June 2020.
- Åman M (1990). Spanska sjukan: den svenska epidemin 1918–1920 och dess internationella bakgrund (in Swedish). Uppsala; Stockholm: Ubsaliensis Academiae; Distributor, Almqvist & Wiksell International. ISBN 978-91-554-2587-6. OCLC 22451542.
- ”Spanish Influenza in Japanese Armed Forces, 1918–1920”. Centers for Disease Control and Prevention (CDC).
- Historical research report from University of Indonesia, School of History, as reported in Emmy Fitri. Looking Through Indonesia’s History For Answers to Swine Flu Archived 2 November 2009 at the Wayback Machine. The Jakarta Globe. 28 October 2009 edition.
- Kohn 2007.
- ”İSPANYOL GRİBİNİN DÜNYA VE OSMANLI DEVLETİ ÜZERİNDEKİ ETKİLERİ” (in Turkish). Archived from the original on 8 November 2020. Retrieved 2 November 2020.
- ”1918 flu centenary: How to survive a pandemic”. Archived from the original on 9 July 2019. Retrieved 19 June 2019.
- Rice G, Bryder L. Black November: the 1918 influenza pandemic in New Zealand (Second, revised and enlarged ed.). Christchurch, NZ. ISBN 978-1-927145-91-3. OCLC 960210402.
- ”1918–1920 – Influenza and Pneumonia Pandemic – Nationwide ~820,000–850,000 – Deadliest American Disasters and Large-Loss-of-Life Events”. Archived from the original on 30 May 2020. Retrieved 22 May 2020.
- The Great Pandemic: The United States in 1918–1919, U.S. Department of Health & Human Services.
- ”The silent invader – Digital Collections – National Library of Medicine”. Archived from the original on 24 July 2020. Retrieved 15 April 2020.
- ”Flu Epidemic Hit Utah Hard in 1918, 1919”. Deseret News. 28 March 1995. Archived from the original on 30 November 2012. Retrieved 7 July 2012.
- ”The Great Pandemic of 1918: State by State”. 5 March 2018. Archived from the original on 6 May 2009. Retrieved 4 May 2009.
- ”A deadly virus rages throughout Canada at the end of the First World War”. CBC History.
- ”A gripe espanhola no Brasil – Elísio Augusto de Medeiros e Silva, empresário, escritor e membro da AEILIJ” (in Portuguese). Jornal de Hoje. Archived from the original on 2 February 2014. Retrieved 22 January 2014.
- ”The ”bird flu” that killed 40 million”. BBC News. 19 October 2005. Archived from the original on 7 November 2017. Retrieved 26 April 2009.
- Hays 1998.
- Marcus H (1996). Haile Sellassie I: The formative years, 1892–1936. Trenton, NJ: Red Sea Press. pp. 36ff.
- Pankhurst 1991, pp. 48f.
- Pankhurst 1991, p. 63.
- Spinney 2018, pp. 167–69.
- ”Viruses of mass destruction”. Fortune. CNN. 1 November 2004. Archived from the original on 6 May 2009. Retrieved 30 April 2009.
- Spinney 2018, p. 134.
- Denoon 2004.
- Wishart S (July–August 2018). ”How the 1918 flu spread”. New Zealand Geographic (152): 23. Archived from the original on 3 August 2018. Retrieved 3 August 2018.
- Afkhami 2003; Afkhami 2012.
- FitzGerald J. ”Coronavirus: forgotten lessons of the Spanish flu pandemic”. The Irish Times. Archived from the original on 5 May 2020. Retrieved 14 June 2020. Yet the ”Spanish” flu epidemic of 1918–19 resulted in about 25,000 extra deaths in Ireland, many of them young adults. This was almost as many deaths as occurred among the Irish fighting in the First World War.
- ”Official Ireland Statistics Office”. Central Statistics Office Ireland. Archived from the original on 14 June 2020. Retrieved 14 June 2020. Total deaths in Ireland 1916: 50,627
- Phillips H. ”South Africa bungled the Spanish flu in 1918. History mustn’t repeat itself for COVID-19”. The Conversation. Archived from the original on 13 June 2020. Retrieved 26 May 2020.
- Spinney 2018, p. 72.
- Pankhurst 1991, pp. 51ff.
- ”Influenza of 1918 (Spanish Flu) and the US Navy”. history.navy.mil. Archived from the original on 11 January 2015. Retrieved 14 May 2009.
- Bell D, Nicoll A, Fukuda K, Horby P, Monto A, Hayden F, et al. (January 2006). ”Non-pharmaceutical interventions for pandemic influenza, international measures”. Emerging Infectious Diseases. 12 (1): 81–7. doi:10.3201/eid1201.051370. PMC 3291414. PMID 16494722.
- Shanks GD, Brundage JF (February 2013). ”Pacific islands which escaped the 1918-1919 influenza pandemic and their subsequent mortality experiences”. Epidemiology and Infection. 141 (2): 353–6. doi:10.1017/S0950268812000866. PMID 22564320. S2CID 23794327. Archived from the original on 11 July 2021. Retrieved 11 July 2021.
- Ryan J, ed. (2009). Pandemic Influenza: Emergency planning and community preparedness. Boca Raton, FL: CRC Press. p. 24.
- ”Colonial Annual Report”, 1919
- Iijima W (2003). ”Spanish influenza in China, 1918–1920: a preliminary probe”. In Phillips H, Killingray D (eds.). The Spanish Flu Pandemic of 1918: New Perspectives. London and New York: Routledge. pp. 101–09.
- Killingray D, Phillips H (2003). The Spanish Influenza Pandemic of 1918–1919: New Perspectives. Routledge. ISBN 978-1-134-56640-2. Archived from the original on 27 July 2021. Retrieved 9 November 2020.
- Killingray D, Phillips H (2003). The Spanish Influenza Pandemic of 1918–1919: New Perspectives. Routledge. ISBN 978-1-134-56640-2. Archived from the original on 27 July 2021. Retrieved 9 November 2020.
- Iijima W (1998). The Spanish influenza in China, 1918–1920. OCLC 46987588.
- Iijima W (1998). The Spanish influenza in China, 1918–1920. Spanish ’Flu 1918–1998: Reflections on the Influenza Pandemic of 1918 after 80 Years. Cape Town, South Africa. Archived from the original on 11 April 2020. Retrieved 11 April 2020.
- Killingray D, Phillips H (2003). The Spanish Influenza Pandemic of 1918–1919: New Perspectives. Routledge. p. 105. ISBN 978-1-134-56640-2. Archived from the original on 27 July 2021. Retrieved 9 November 2020.
- Alita XP, Ling T, Bin ZH, Xiao-ting LI (2010). ”Epidemic analysisi of 1918 Spanish influenza in China based on literatures”. Journal of Medical Informatics. 31 (1): 47–50.
- Hong NH (2015). ”Epidemic Analysis of 1918 Influenza in China – Research on Huolu County in Zhili Province”. Historical Research in Auhui.
- Simonsen et al. 1998.
- Hanssen, Olav. Undersøkelser over influenzaens optræden specielt i Bergen 1918–1922. Bg. 1923. 66 s. ill. (Haukeland sykehus. Med. avd. Arb. 2) (Klaus Hanssens fond. Skr. 3)
- Knobler SL, Mack A. ”Institute of Medicine (US) Forum on Microbial Threats”. NCBI. Retrieved 17 August 2021.
- Barry 2004, p. 239.
- ”Key Facts about Swine Influenza”. Archived from the original on 4 May 2009. Retrieved 30 April 2009.
- Spinney 2018, pp. 180–82.
- Spinney 2018, pp. 183–84.
- Mamelund SE (February 2006). ”A socially neutral disease? Individual social class, household wealth and mortality from Spanish influenza in two socially contrasting parishes in Kristiania 1918–19”. Social Science & Medicine. 62 (4): 923–40. doi:10.1016/j.socscimed.2005.06.051. PMID 16084634.
- He et al. 2011.
- He et al. 2013.
- ”The ’Spanish’ Influenza pandemic and its relation to World War I”. World War I Centenary. University of Oxford JISC. Archived from the original on 5 August 2020. Retrieved 11 August 2020.
- Garret 2007.
- Lindley R. ”The Forgotten American Pandemic: Historian Dr. Nancy K. Bristow on the Influenza Epidemic of 1918”. hnn.us. Archived from the original on 8 August 2014. Retrieved 4 August 2014.
- ”What can the Spanish Flu teach us about the COVID-19 pandemic?”. World Economic Forum. 2 April 2020. Archived from the original on 9 April 2020. Retrieved 8 April 2020.
- Correia S, Luck S, Verner E (2020). ”Pandemics Depress the Economy, Public Health Interventions Do Not: Evidence from the 1918 Flu”. SSRN Working Paper Series. doi:10.2139/ssrn.3561560. ISSN 1556-5068. S2CID 216202133. Archived from the original on 27 July 2021. Retrieved 14 April 2020.
- Kemp, John (7 April 2020). ”RPT-COLUMN-Coronavirus lockdowns: can we learn from the 1918 influenza pandemic? Kemp”. Reuters. Retrieved 17 August 2021.
- Almond D (1 August 2006). ”Is the 1918 Influenza Pandemic Over? Long‐Term Effects of In Utero Influenza Exposure in the Post‐1940 U.S. Population”. Journal of Political Economy. 114 (4): 672–712. doi:10.1086/507154. ISSN 0022-3808. S2CID 20055129.
- Beach B, Ferrie JP, Saavedra MH (2018). ”Fetal shock or selection? The 1918 influenza pandemic and human capital development”. nber.org. doi:10.3386/w24725. Archived from the original on 18 June 2018. Retrieved 18 June 2018.
- Vilensky, Foley & Gilman 2007.
- Fottrell Q (25 August 2020). ”Another warning from the 1918 flu for COVID-19: ’Survival does not mean that individuals fully recovered'”. MarketWatch. Archived from the original on 24 August 2020.
- Honigsbaum 2008.
- Morrisey 1986.
- Benedict & Braithwaite 2000, p. 38.
- Crosby 2003, pp. 320–22.
- Ball S (13 February 2012). ”Downton Abbey, Season Two, Episode Six Recap: Nobody Expects the Spanish Influenza!”. Vanity Fair. Archived from the original on 23 October 2020. Retrieved 9 December 2020.
- Meyer R (29 April 2016). ”Human extinction isn’t that unlikely”. The Atlantic. Archived from the original on 1 May 2016. Retrieved 6 February 2018.
- ”WHO Coronavirus Disease (COVID-19) Dashboard”. WHO. Archived from the original on 16 April 2020. Retrieved 27 February 2021.
- Hilleman MR (August 2002). ”Realities and enigmas of human viral influenza: pathogenesis, epidemiology and control”. Vaccine. 20 (25–26): 3068–87. doi:10.1016/S0264-410X(02)00254-2. PMID 12163258.
- Potter CW (October 2001). ”A history of influenza”. Journal of Applied Microbiology. 91 (4): 572–9. doi:10.1046/j.1365-2672.2001.01492.x. PMID 11576290.
- Biggerstaff M, Cauchemez S, Reed C, Gambhir M, Finelli L (September 2014). ”Estimates of the reproduction number for seasonal, pandemic, and zoonotic influenza: a systematic review of the literature”. BMC Infectious Diseases. 14 (1): 480. doi:10.1186/1471-2334-14-480. PMC 4169819. PMID 25186370.
- Valleron AJ, Cori A, Valtat S, Meurisse S, Carrat F, Boëlle PY (May 2010). ”Transmissibility and geographic spread of the 1889 influenza pandemic”. Proceedings of the National Academy of Sciences of the United States of America. 107 (19): 8778–81. Bibcode:2010PNAS..107.8778V. doi:10.1073/pnas.1000886107. PMC 2889325. PMID 20421481.
- Mills CE, Robins JM, Lipsitch M (December 2004). ”Transmissibility of 1918 pandemic influenza”. Nature. 432 (7019): 904–6. Bibcode:2004Natur.432..904M. doi:10.1038/nature03063. PMC 7095078. PMID 15602562.
- Taubenberger JK, Morens DM (January 2006). ”1918 Influenza: the mother of all pandemics”. Emerging Infectious Diseases. 12 (1): 15–22. doi:10.3201/eid1201.050979. PMC 3291398. PMID 16494711.
- ”Report of the Review Committee on the Functioning of the International Health Regulations (2005) in relation to Pandemic (H1N1) 2009” (PDF). 5 May 2011. p. 37. Archived (PDF) from the original on 14 May 2015. Retrieved 1 March 2015.
- Spreeuwenberg P, Kroneman M, Paget J (December 2018). ”Reassessing the Global Mortality Burden of the 1918 Influenza Pandemic”. American Journal of Epidemiology. 187 (12): 2561–2567. doi:10.1093/aje/kwy191. PMID 30202996.
- Morens DM, Fauci AS (April 2007). ”The 1918 influenza pandemic: insights for the 21st century”. The Journal of Infectious Diseases. 195 (7): 1018–28. doi:10.1086/511989. PMID 17330793.
- Johnson NP, Mueller J (2002). ”Updating the accounts: global mortality of the 1918-1920 ”Spanish” influenza pandemic”. Bulletin of the History of Medicine. 76 (1): 105–15. doi:10.1353/bhm.2002.0022. PMID 11875246.
- Lin II R, Karlamangla S (6 March 2020). ”Why the coronavirus outbreak isn’t likely to be a repeat of the 1918 Spanish flu”. Los Angeles Times.
- Schwarzmann SW, Adler JL, Sullivan RJ, Marine WM (June 1971). ”Bacterial pneumonia during the Hong Kong influenza epidemic of 1968-1969”. Archives of Internal Medicine. 127 (6): 1037–41. doi:10.1001/archinte.1971.00310180053006. PMID 5578560.
- Michaelis M, Doerr HW, Cinatl J (August 2009). ”Novel swine-origin influenza A virus in humans: another pandemic knocking at the door”. Medical Microbiology and Immunology. 198 (3): 175–83. doi:10.1007/s00430-009-0118-5. PMID 19543913. S2CID 20496301.
- Donaldson LJ, Rutter PD, Ellis BM, Greaves FE, Mytton OT, Pebody RG, Yardley IE (December 2009). ”Mortality from pandemic A/H1N1 2009 influenza in England: public health surveillance study”. BMJ. 339: b5213. doi:10.1136/bmj.b5213. PMC 2791802. PMID 20007665.
- ”First Global Estimates of 2009 H1N1 Pandemic Mortality Released by CDC-Led Collaboration”. Centers for Disease Control and Prevention (CDC). 25 June 2012. Retrieved 7 July 2012.
- Kelly H, Peck HA, Laurie KL, Wu P, Nishiura H, Cowling BJ (5 August 2011). ”The age-specific cumulative incidence of infection with pandemic influenza H1N1 2009 was similar in various countries prior to vaccination”. PLOS One. 6 (8): e21828. Bibcode:2011PLoSO…621828K. doi:10.1371/journal.pone.0021828. PMC 3151238. PMID 21850217.
- Dawood FS, Iuliano AD, Reed C, Meltzer MI, Shay DK, Cheng PY, et al. (September 2012). ”Estimated global mortality associated with the first 12 months of 2009 pandemic influenza A H1N1 virus circulation: a modelling study”. The Lancet. Infectious Diseases. 12 (9): 687–95. doi:10.1016/S1473-3099(12)70121-4. PMID 22738893.
- Riley S, Kwok KO, Wu KM, Ning DY, Cowling BJ, Wu JT, et al. (June 2011). ”Epidemiological characteristics of 2009 (H1N1) pandemic influenza based on paired sera from a longitudinal community cohort study”. PLoS Medicine. 8 (6): e1000442. doi:10.1371/journal.pmed.1000442. PMC 3119689. PMID 21713000.
- Wong JY, Kelly H, Ip DK, Wu JT, Leung GM, Cowling BJ (November 2013). ”Case fatality risk of influenza A (H1N1pdm09): a systematic review”. Epidemiology. 24 (6): 830–41. doi:10.1097/EDE.0b013e3182a67448. PMC 3809029. PMID 24045719.
- ”WHO Europe – Influenza”. World Health Organization (WHO). June 2009. Archived from the original on 17 June 2009. Retrieved 12 June 2009.
- CDC (28 October 2019). ”Key Facts About Influenza (Flu)”. citing Tokars, Olsen& Reed (2018). cdc.gov. Retrieved 10 March 2020.
- Tokars JI, Olsen SJ, Reed C (May 2018). ”Seasonal Incidence of Symptomatic Influenza in the United States”. Clinical Infectious Diseases. 66 (10): 1511–1518. doi:10.1093/cid/cix1060. PMC 5934309. PMID 29206909.
- ”Influenza: Fact sheet”. World Health Organization (WHO). 6 November 2018. Archived from the original on 17 December 2019. Retrieved 25 January 2020.
- ”H1N1 fatality rates comparable to seasonal flu”. The Malaysian Insider. Washington, D.C., USA. Reuters. 17 September 2009. Archived from the original on 20 October 2009. Retrieved 26 September 2009.
- ”Open Collections Program: Contagion, Spanish Influenza in North America, 1918–1919”. Archived from the original on 20 November 2016. Retrieved 22 November 2016.
- Harder TC, Ortrud W. ”Chapter Two: Avian Influenza”. Influenza Report 2006. published online. Archived from the original on 9 August 2017. Retrieved 26 October 2007. Sometimes a virus contains both avian-adapted genes and human-adapted genes. Both the H2N2 and H3N2 pandemic strains contained avian flu virus RNA segments. ”While the pandemic human influenza viruses of 1957 (H2N2) and 1968 (H3N2) arose through reassortment between human and avian viruses, the influenza virus causing the ’Spanish flu’ in 1918 appears to be entirely derived from an avian source (Belshe 2005).
- Taubenberger et al. 2005.
- Antonovics, Hood & Baker 2006.
- Vana & Westover 2008.
- dos Reis, Hay & Goldstein 2009.
- ”Researchers reconstruct 1918 pandemic influenza virus; effort designed to advance preparedness”. Center for Disease Control. Archived from the original on 19 October 2011. Retrieved 2 September 2009.
- ”Closing In On a Killer: Scientists Unlock Clues to the Spanish Influenz Virus”. National Museum of Health and Medicine. Archived from the original on 2 May 2020. Retrieved 23 March 2020.
- Kolata G (16 February 1999). ”Scientists Uncover Clues To Flu Epidemic of 1918”. The New York Times. Archived from the original on 22 March 2020. Retrieved 23 March 2020.
- Kobasa & et al. 2007.
- ”Research on monkeys finds resurrected 1918 flu killed by turning the body against itself”. USA Today. Archived from the original on 5 March 2009. Retrieved 14 August 2008.
- ”Body exhumed in fight against flu”. BBC News. Archived from the original on 17 September 2008. Retrieved 16 September 2008.
- In Search of Spanish Flu (documentary). BBC Four.
- Fox 2008.
- Fox 2010.
- Crosby 1976, p. 295.
- Madhav 2013.
- Branswell H (5 December 2018). ”A shot-in-the-dark e-mail leads to a century-old family treasure – and hope of cracking a deadly flu’s secret”. Stat. Archived from the original on 5 December 2018. Retrieved 5 December 2018.
- Hammond JA, Rolland W, Shore TH (14 July 1917). ”Purulent bronchitis: A study of cases occurring amongst the British troops at a base in France”. The Lancet. 190 (4898): 41–45. doi:10.1016/S0140-6736(01)56229-7. Archived from the original on 27 July 2021. Retrieved 8 December 2018. PDF version here Archived 26 August 2019 at the Wayback Machine
- ”The Influenza Pandemic of 1918”. World War 1 Centenary. Oxford, England: University of Oxford. Archived from the original on 9 December 2018. Retrieved 8 December 2018.
- Yu, X.; Tsibane, T.; McGraw, P. A.; House, F. S.; Keefer, C. J.; Hicar, M. D.; Tumpey, T. M.; Pappas, C.; Perrone, L. A.; Martinez, O.; Stevens, J.; Wilson, I. A.; Aguilar, P. V.; Altschuler, E. L.; Basler, C. F.; Crowe Jr, J. E. (2008). ”Neutralizing antibodies derived from the B cells of 1918 influenza pandemic survivors”. Nature. 455 (7212): 532–536. Bibcode:2008Natur.455..532Y. doi:10.1038/nature07231. PMC 2848880. PMID 18716625. Retrieved 24 October 2021.
- Sawchuk LA (August 2009). ”Brief communication: Rethinking the impact of the 1918 influenza pandemic on sex differentials in mortality”. American Journal of Physical Anthropology. 139 (4): 584–90. doi:10.1002/ajpa.21022. PMID 19280673. Archived from the original on 27 July 2021. Retrieved 10 December 2020.
- Noymer A, Garenne M (September 2000). ”The 1918 influenza epidemic’s effects on sex differentials in mortality in the United States”. Population and Development Review. 26 (3): 565–81. doi:10.1111/j.1728-4457.2000.00565.x. PMC 2740912. PMID 19530360.
- Paskoff T, Sattenspiel L (January 2019). ”Sex- and age-based differences in mortality during the 1918 influenza pandemic on the island of Newfoundland”. American Journal of Human Biology. 31 (1): e23198. doi:10.1002/ajhb.23198. PMID 30488509. S2CID 54122685. Archived from the original on 19 October 2020. Retrieved 10 December 2020.
- Rewegan A, Bogaert K, Yan M, Gagnon A, Herring DA (10 September 2015). ”The first wave of the 1918 influenza pandemic among soldiers of the Canadian expeditionary force”. American Journal of Human Biology. 27 (5): 638–45. doi:10.1002/ajhb.22713. PMID 25820782. S2CID 11033776. Archived from the original on 27 July 2021. Retrieved 10 December 2020.








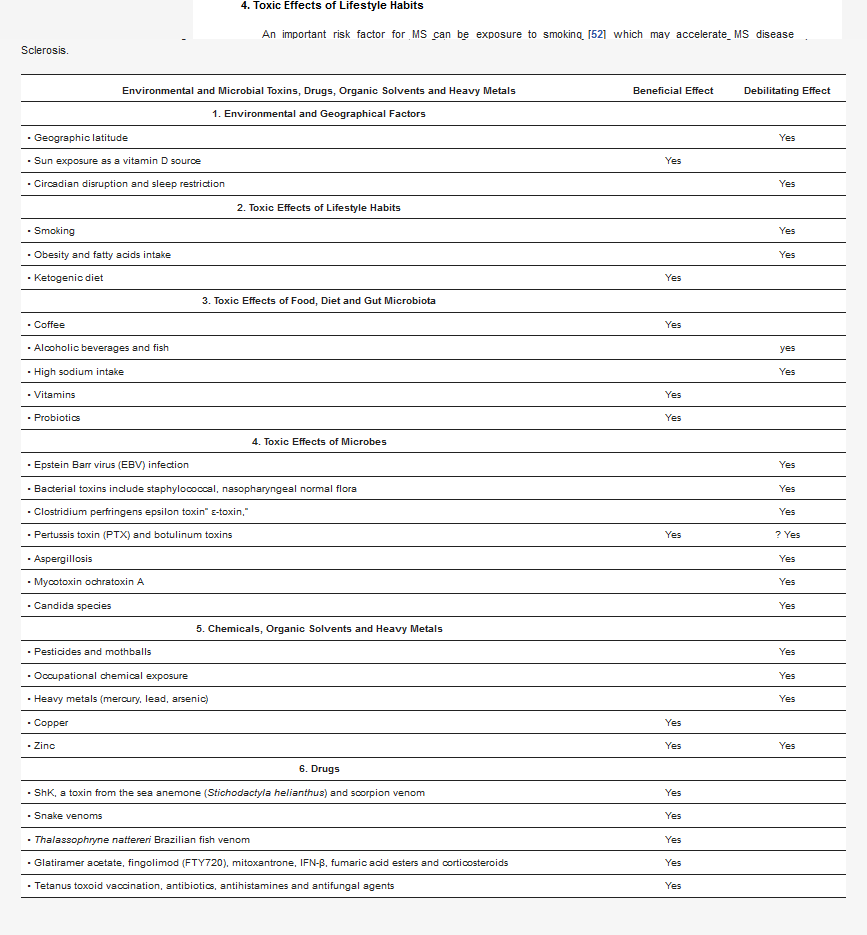


















 Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/
Lähde: https://www.hindawi.com/journals/jnme/2018/7195760/